

Vorplanarisierte Triphenylamin-basierte lineare gemischtvalente Ladungstransfersysteme
Professor Fred Wudl zum 80. Geburtstag gewidmet
Abstract
Drei lineare Dimere mit jeweils zwei redoxaktiven planarisierten Triphenylaminen wurden synthetisiert, und ihre Struktur wurde mittels Röntgenstrukturanalyse aufgelöst. Diese Verbindungen sind von großem Interesse, da ihre Radikalkationen einen Selbstaustausch des ungepaarten Elektrons zwischen den beiden Redoxzentren aufweisen. Dieser Prozess wurde eingehend mittels Elektronenspinresonanz-Spektroskopie, Absorptionsspektroskopie und (zeitabhängigen) Dichtefunktionaltheorie-Rechnungen untersucht. Ein Vergleich der Schlüsselparameter des Elektronentransfers mit nicht-planarisierten stickstoffzentrierten Bausteinen unterstreicht den Einfluss von Redoxzentren mit niedrigen inneren Reorganisationsenergien. Hingegen bleibt der entfernungsabhängige Abschwächungsfaktor des Superaustauschmechanismus ähnlich.
Einleitung
Elektronentransferprozesse werden weitestgehend von zwei grundlegenden Parametern bestimmt; der elektronischen Kopplung V zwischen dem Elektronendonor und dem Elektronenakzeptor sowie der Reorganisationsenergie λ, welche sich aus inter- und intramolekularen Beiträgen zusammensetzt.1, 2 Gemischtvalente Ladungstransfersysteme (MV-CT) mit mindestens zwei chemisch äquivalenten Einheiten, aber unterschiedlichen Ladungen, wurden entwickelt und untersucht, um das Zusammenspiel der Faktoren, die den Elektronentransfer steuern, zu verstehen.3, 4
Robin und Day schlugen eine Klassifizierung von MV-CT-Systemen anhand der elektronischen Wechselwirkungsstärke zwischen den redoxaktiven Zentren vor.5 Klasse I ist der Extremfall der Diabatizität, bei dem die Redoxzentren aufgrund vernachlässigbarer elektronischer Kommunikation unabhängig voneinander sind. Klasse-II-Systeme hingegen weisen eine hinreichende elektronische Kommunikation zwischen den Redoxeinheiten auf. Dies äußert sich z. B. in der Aufspaltung der Redoxpotentiale. Bemerkenswert dabei ist, dass sich die Ladung auf einer der beiden spezifischen Redoxeinheiten befindet, jedoch durch einen thermisch oder optisch induzierten Elektronentransfer übertragen werden kann. Eine Potentialkurve mit zwei Minima entlang der Elektronentransferkoordinate ist typisch für Klasse-II-Verbindungen. In Klasse III zwingt eine außerordentlich starke Kopplung die Ladung zur Delokalisierung über das gesamte System. Klasse-III-Verbindungen werden auch als “Ladungsresonanzsysteme” bezeichnet und weisen eine Potentialkurve mit einem einzigen Minimum entlang der Elektronentransferkoordinate auf.
 max [Gl. 1].
max [Gl. 1].
 (1)
(1) (2)
(2) (3)
(3) (4)
(4) (5)
(5)Dabei ist μeg das Übergangsdipolmoment, welches sich durch Integration der IV-CT-Bande berechnen lässt. Δμ12 ist die diabatische Dipolmomentdifferenz, die sich aus μeg und Δμeg, der adiabatischen Dipolmomentdifferenz, ergibt. Letztere wird genähert durch den Abstand der Redoxzentren r multipliziert mit der Elementarladung e.
Molekulare MV-CT-Systeme weisen zwei oxidierbare oder reduzierbare Bausteine auf, welche kovalent durch molekulare Brücken verbunden sind. Die molekularen Brücken erfüllen zwei Zwecke. Sie fungieren als elektronisches Koppelmedium und als Distanzhalter. Typischerweise sind sie aus Kohlenstoffgerüsten, wie Aromaten,8-11 Alkinen,8, 12 Cyclophanen,13-15 Oligo-p-phenylvinylenen16, 17 usw., oder Kombinationen davon aufgebaut.18, 19 Diese molekularen Brücken ermöglichen eine Feinabstimmung der elektronischen Kopplung sowohl bei kurzen als auch bei langen Distanzen.
Triarylamine gehören hierbei zu den am besten untersuchten Redoxzentren für MV-CT-Systeme.3, 4 Wichtige Eigenschaften dieser Redoxzentren sind eine reversible Oxidation bei niedrigen Potentialen und eine hinreichende Stabilität der jeweiligen Radikalkationen. In Triphenylamin-MV-CT-Systemen sind die IV-CT-Banden typischerweise gut von lokalen Absorptionsbanden getrennt, was die Analyse der Bandenform vereinfacht. Jedoch neigen die propellerförmigen Triarylamine dazu, bei der Oxidation zu planarisieren, was zu größeren inneren Reorganisationsenergien führt. Um dieses Problem zu beheben, haben wir drei verschiedene Dimere aus bereits vorplanarisierten Triarylaminen (N-Heterotriangulenen (N-HTAs))20-23 als Redoxzentren und Acetylen (DTA), p-Phenylen (DTB) und Tolan (DTT) als molekulare Brücken entworfen (Abbildung 1).

Chemische Strukturen der N-Heterotriangulen-Dimere DTA, DTB und DTT.
Fang et al. haben bereits gezeigt, dass das Oxidationspotential des unsubstituierten N-HTA in CH2Cl2 (+0.34 V vs. Fc/Fc+) niedriger ist als das von Triphenylamin (+0.54 V vs. Fc/Fc+).24 Das bei der Oxidation erzeugte Radikalkation des N-HTA erweist sich zudem als etwas beständiger als das von Triphenylamin.24, 25 Um den Einfluss der Planarisierung auf die Reorganisationsenergie und die elektronische Kopplung zu bewerten, untersuchten wir die Dimere und deren Radikalkationen mittels elektrochemischer Methoden, Absorptionsspektroskopie und Elektronenspinresonanz-(EPR-)Spektroskopie. Die IV-CT-Parameter wurden durch Analyse der Absorptionsbanden gemäß der Mulliken-Hush-Theorie bestimmt. Die experimentellen Ergebnisse wurden durch Berechnungen mit dem Dichtefunktional BLYP35, das 35 % exakten Hartree-Fock-(HF-)Austausch in das BLYP-Austauschkorrelationsfunktional mischt, weiter aufgeklärt. Es hat sich gezeigt, dass dieses Funktional gute Ergebnisse für Fragestellungen liefert, bei denen die Lokalisierung/Delokalisierung von Ladungen von Bedeutung ist.26, 27
Ergebnisse und Diskussion
Die unterschiedlich verknüpften Dimere DTA, DTB und DTT wurden aus bekannten N-HTA-Bausteinen und ausgewählten molekularen Brücken mittels übergangsmetallkatalysierter Kreuzkupplungsreaktionen synthetisiert. Die Dimere sind stabile, farblose bis blassgelbe Feststoffe, welche in gängigen organischen Lösungsmitteln löslich sind. Die synthetischen Details und die Charakterisierungsdaten sind in der SI, Abschnitt 3, zu finden.
Für die Röntgenkristallographie geeignete Einkristalle des DTA und DTT wurden durch langsame Flüssigkeitsdiffusion von MeOH in CH2Cl2-Lösungen der Verbindungen bei Raumtemperatur erhalten, während Einkristalle des DTB durch langsames Verdampfen von CD2Cl2 erhalten wurden (Abbildung 2). Obwohl aufgrund der Acetylenbrücke eine koplanare Orientierung der zwei N-HTAs erwartet wird, zeigt das DTA eine verzerrte Struktur, bei der die beiden N-HTAs entlang der linearen C(sp)-Brücke mit einem Torsionswinkel Χ von 46.8° zueinander verdreht sind. Dennoch zeigen beide N-HTAs eine charakteristische Planarisierung um das Stickstoffzentrum mit einer Summe der C-N-C/(α,β,γ)-Winkel von 358.1°. Für das DTA wird im festen Zustand kein spezifisches Packungsmotiv beobachtet (Details der Kristallpackung sind in der SI, Abschnitt 5, zu finden). Im Gegensatz dazu sind die über Benzolbrücken verbundenen N-HTAs des DTB aufgrund der kristallographischen Inversionssymmetrie koplanar angeordnet. Zudem wird ein Torsionswinkel Χ von 34.3° zwischen den N-HTAs und der Phenylbrücke beobachtet. Auch im DTB sind die Stickstoffzentren der N-HTAs nahezu planar. Analog zum DTB sind die N-HTAs im DTT durch die Tolanbrücke ebenfalls koplanar verbunden. Auch hier stellen wir aufgrund eines Inversionssymmetriezentrums eine fehlende “globale Torsion” fest. Zwischen den N-HTAs und den benachbarten Phenylringen der Tolanbrücke wird ein Torsionswinkel Χ von 44.4° beobachtet. Außerdem wird aus Symmetriegründen keine Torsion zwischen den an den Acetylenrest angrenzenden Phenylgruppen beobachtet, was ein Merkmal des unsubstituierten Tolans ist.28 Eine charakteristische Planarisierung um die Stickstoffzentren der N-HTAs mit einer Summe der C-N-C/(α,β,γ)-Winkel von 359.8° wird für das DTT gefunden. Dies ist konsistent mit den Beobachtungen für das DTA und DTB. Insgesamt wird die Anordnung höchstwahrscheinlich durch die Packung im Kristall erzwungen.

ORTEP-Diagramm des DTA (oben), des DTB (Mitte) und des DTT (unten; thermische Ellipsoide mit 50 % Wahrscheinlichkeit; Wasserstoffe sind nicht dargestellt).36 Übersicht der vorhandenen Torsionswinkel Χ und ∑C-N-C/(α, β, γ)-Winkel.
Die Differenz der Oxidationspotentiale im Square-Wave-Voltammogramm (SWV, Abbildung S22) erlaubt eine qualitative Abschätzung der elektronischen Interaktion der N-HTAs. Das SWV von DTA in 0.2 m TBAPF6/CH2Cl2-Lösung zeigt zwei gut aufgelöste Oxidationssignale bei +0.37 und +0.54 V vs. Fc/Fc+. Für DTB liegen die Oxidationspotentiale bei +0.38 und +0.48 V vs. Fc/Fc+, wodurch der Abstand auf der Potentialskala deutlich kleiner ausfällt. Für das Tolan-verbrückte DTT verringert sich die Differenz der Oxidationspotentiale, welche als +0.39 und +0.40 V vs. Fc/Fc+ bestimmt wurden, sogar auf nur 0.01 V. In Tabelle 1 sind die Oxidationspotentiale, deren Differenz und die daraus abgeleitete Komproportionierungskonstante zusammengefasst. Dadurch lässt sich bereits feststellen, dass die elektronische Kommunikation in DTA am ausgeprägtesten ist und über DTB zu DTT stetig abnimmt.
Verbindung |
Eox1 |
Eox2 |
ΔE [V] |
KCO |
|---|---|---|---|---|
DTA |
+0.37 |
+0.54 |
0.17 |
840 |
DTB |
+0.38 |
+0.48 |
0.10 |
53 |
DTT |
+0.39 |
+0.40 |
0.01 |
1.5 |
Eine weitere Möglichkeit, die elektronische Kopplung zu bewerten, besteht in Orbitalinteraktionsdiagrammen (siehe SI Abschnitt 8.2.3). Der energetische Unterschied zwischen HOMO und HOMO−1 liefert eine grobe Einschätzung der elektronischen Kopplung, welche selbst wiederum eine Folge der Orbitalwechselwirkungen zwischen den Elektronendonoren sowie deren Verbindungsbrücke ist.29, 30 Diese Differenz liegt für DTA bei 0.42 eV und nimmt über DTB mit 0.20 eV zu DTT mit 0.12 eV stetig ab und spiegelt somit den Trend der elektrochemischen Untersuchung wider.
Die Bestimmung von elektronischer Kopplung und Reorganisationsenergie kann durch weitere Dichtefunktionaltheorie-(DFT-)Rechnungen weiter verfeinert werden (Abbildung 3).29, 31 Dazu wurden unsymmetrische und symmetrische Strukturen berechnet, welche die lokalisierten elektronischen Strukturen und den delokalisierten Übergangszustand, der bei thermisch angeregtem Elektronentransfer durchschritten werden muss, beschreiben.

Energiediagramme des Intervalenz-Ladungstransfers für DTA.+ (oben), DTB.+ (Mitte) und DTT.+ (unten). Die unsymmetrischen Spindichten entsprechen der lokalisierten Ladungsverteilung und die symmetrischen dem delokalisierten Übergangszustand (Isofläche: ±0.001 a.u.). Es sind Ergebnisse aus TD-BLYP35-Berechnungen in CH2Cl2 gezeigt.
Die Reorganisationsenergie der Mulliken-Hush-Theorie kann dabei aus zeitabhängigen (TD-)DFT-Rechnungen extrahiert werden. Sie entspricht der Anregungsenergie des niederenergetischsten Übergangs der unsymmetrischen Struktur. Eine Betrachtung der natürlichen Übergangsorbitale (NTOs) beweist dabei, dass der gewählte Übergang einem Ladungstransfer entspricht (SI Abschnitt 8.3). Das Start-NTO befindet sich auf dem neutralen N-HTA, während das Ziel-NTO auf dem oxidiertem N-HTA liegt. Als Reorganisationsenergie wurden 4479 cm−1 für DTA.+, 5468 cm−1 für DTB.+ und 6152 cm−1 für DTT.+ berechnet.
Die Aktivierungsbarriere lässt sich aus der Energiedifferenz der unsymmetrischen und symmetrischen Struktur bestimmen. Für DTA.+ liegt diese bei 0.3 kJ mol−1 sowie 5.3 kJ mol−1 für DTB.+ und 13.4 kJ mol−1 für DTT.+.
Eine Berechnung der elektronischen Kopplung erfolgt über die Energie des niedrigsten elektronischen Übergangs der symmetrischen Übergangszustandsstruktur. Diese Energie entspricht dem doppelten Zahlenwert der elektronischen Kopplung. Daraus errechnet sich eine elektronische Kopplung von 1796 cm−1 für DTA.+ sowie 1054 cm−1 für DTB.+ und 410 cm−1 für DTT.+.
Für alle N-HTA-Dimere ist die lokalisierte Struktur am stabilsten. Folglich lassen sich diese Systeme als Robin-Day-Klasse II einstufen. Das DTA.+ ist allerdings ein Grenzfall. Auch wenn sich der Großteil der Spindichte auf einem N-HTA zentriert, so finden sich auch Anteile davon auf dem zweiten N-HTA. Diese Beobachtung deutet darauf hin, dass die positive Ladung ebenfalls etwas delokalisiert ist, und unterstreicht die starke elektronische Kommunikation, welche durch den Acetylen-Linker vermittelt wird.
Im Weiteren wurde eine Lokalisierung bzw. Delokalisierung der positiven Ladung mittels EPR-Spektroskopie untersucht. Die temperaturabhängigen CW-X-Band-EPR-Messungen wurden direkt nach der In-situ-Oxidation mittels SbCl5 der Dimere in 1 mm CH2Cl2-Lösung durchgeführt. Bei 95 K wurden für alle Systeme isotrope Signale gefunden. Die ermittelten g-Werte sind dabei g =2.0016, g
=2.0016, g =2.0043 und g
=2.0043 und g =2.0015 und die Linienbreiten W
=2.0015 und die Linienbreiten W =0.720×10−4 cm−1/GHz, W
=0.720×10−4 cm−1/GHz, W =0.774×10−4 cm−1/GHz und W
=0.774×10−4 cm−1/GHz und W =0.290×10−4 cm−1/GHz.
=0.290×10−4 cm−1/GHz.
Bei niedrigen Temperaturen wurde eine Triplettaufspaltung des Signals erwartet. Allerdings ist bei tiefen Temperaturen die Linienbreite, aufgrund verschiedener Beiträge,32 größer als die Hyperfeinaufspaltung, was deren Beobachtung verhindert. Bei höheren Temperaturen wird eine Quintettaufspaltung beobachtet (Abbildung 4), welche eine Superhyperfeinkopplung des ungepaarten Elektrons mit zwei äquivalenten 14N-Kernen (14N, I=1, 99.6 % nat. Häufigkeit) impliziert. Als Superhyperfeinkopplungskonstanten wurden A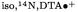 =3.73×10−4 cm−1, A
=3.73×10−4 cm−1, A =3.99×10−4 cm−1 und A
=3.99×10−4 cm−1 und A =4.10×10−4 cm−1 bei RT bestimmt. Die direkte Ausbildung der Quintettaufspaltung bei Temperaturerhöhung legt nahe, dass der Spinaustausch schneller als die Zeitauflösung des EPR-Geräts erfolgt und deswegen ein zeitlich gemitteltes Signal erhalten wird.
=4.10×10−4 cm−1 bei RT bestimmt. Die direkte Ausbildung der Quintettaufspaltung bei Temperaturerhöhung legt nahe, dass der Spinaustausch schneller als die Zeitauflösung des EPR-Geräts erfolgt und deswegen ein zeitlich gemitteltes Signal erhalten wird.

Temperaturabhängige CW-X-Band-EPR-Spektren von DTA.+, DTB.+ und DTT.+ in 1 mm CH2Cl2-Lösung. Experimentelle Bedingungen: Mikrowellenfrequenz ν=8.959 GHz, Modulationsbreite=0.1; 0.01 mT, Mikrowellenleistung=1.00 mW, Modulationsfrequenz=100 kHz, Zeitkonstante=0.1 s. Nähere Informationen und Simulationen befinden sich in der SI.
Da die DFT-Rechnungen DTA.+ als Grenzfall zwischen Robin-Day-Klasse II und III erkennen lassen, führten wir für eine eindeutige Klassifizierung zusätzliche spektroelektrochemische Messungen durch. Dabei wurde DTA elektrochemisch oxidiert, während gleichzeitig die dabei auftretenden Änderungen im IR-Spektrum aufgezeichnet wurden (SI Abbildung S30). Neben der stark ausgeprägten Flanke der IV-CT-Bande unterhalb von 3500 nm zeichnet sich eine scharfe, weniger intensive Absorption bei 4671 nm (2141 cm−1) ab. Diese lässt sich der Anregung der Streckschwingung der zentralen Dreifachbindung zuweisen. Ihr Auftauchen kann als Beweis für eine unsymmetrische Ladungsverteilung im Monokation herangezogen werden, womit DTA.+ zweifelsfrei der Robin-Day-Klasse II zugeordnet werden kann. Aufgrund der Symmetrieauswahlregeln fehlt diese Bande in den IR-Absorptionsspektren des ungeladenen DTA und dessen Dikations. Beide Systeme haben symmetrische Ladungsverteilungen. Analog dazu findet sich im IR-Spektrum von DTT.+ (Abbildung S34) eine ähnliche, aber weitaus weniger intensive Bande bei 4523 nm (2211 cm−1). Auch hier zeigen weder DTT noch DTT2+ Absorptionen in dieser Region. Das DTB.+, welches keine C-C-Dreifachbindung enthält, weist keine Signaturen in oben genannten Bereichen auf (Abbildung S32). Alle Kationen zeigen zusätzlich Schwingungsabsorptionen im Bereich von 5960 bis 6300 nm, welche zu Schwingungen der aromatischen Ringsysteme gehören. Die Zuordnung der Absorptionsbanden zu den entsprechenden Schwingungen erfolgte mittels DFT-Kalkulationen.
Eine experimentelle Quantifizierung des Elektronentransfers kann über die Mulliken-Hush-Analyse der nIR-Absorptionsbanden durchgeführt werden. SbCl5 dient dabei als Oxidationsmittel, um DTA, DTB und DTT in CH2Cl2 stufenweise zum Mono- und anschließend zum Dikation zu oxidieren. Die Umsetzung wurde dabei mittels Absorptionsspektroskopie verfolgt. Die dabei erhaltenen Absorptionsspektren der einzelnen Spezies sind in Abbildung 5 dargestellt. Alle Verbindungen im einfach oxidierten Zustand weisen breite Absorptionsbanden im nIR auf und passen gut zu den berechneten TDDFT-Spektren (SI Abschnitt 8.3). Dementsprechend kann der elektronische Übergang, welcher durch die nIR-Absorption angeregt wird, gemäß der NTO-Analyse dem IV-CT zugeordnet werden. Bei höheren Energien, im sichtbaren Bereich, finden sich hingegen Anregungen lokaler elektronischer Übergänge, jeweils auf dem neutralen oder oxidiertem N-HTA.

Absorptionsspektren von ungeladenem (durchgezogene Linie), kationischem (Strich-Punkte) und dikationischem (Punkte) DTA (oben), DTB (Mitte) und DTT (unten) in CH2Cl2. Die geladenen Spezies wurden in situ durch die Zugabe von SbCl5 generiert.
Die Maxima der nIR-Absorptionsbanden wurden aus Anpassung der experimentellen Daten extrahiert. Diese ermöglichen zudem eine weitere Auswertung mittels Mulliken-Hush-Analyse (SI Abschnitt 7.3). Allerdings waren unsymmetrische Ausgleichkurven notwendig, um die nIR-Absorptionsspektren von DTA.+ und DTB.+ zu beschreiben. Deren Maxima liegen bei 2411 nm (4147 cm−1) für DTA.+ und 2228 nm (4489 cm−1) DTB.+. An dieser Stelle ist anzumerken, dass die nIR-Absorption von DTT.+ teilweise von Absorptionen lokaler Anregungen überlagert wird. Aus diesem Grund haben wir einen ausgewählten Teil des Spektrums mit Gauß-Funktionen angepasst. Gemäß den TDDFT-Berechnungen lässt sich die langwelligste Absorption bei 1399 nm (7148 cm−1) dem IV-CT zuordnen.
Über Gleichungen (1) bis (5) lässt sich nun die Reorganisationsenergie und die elektronische Kopplung für DTA.+, DTB.+ und DTT.+ (Tabelle 2 und Tabelle S5) bestimmen. Die elektronische Kopplung nimmt von 825 cm−1 für DTA.+ über 566 cm−1 für DTB.+ zu 261 cm−1 für DTT.+ stetig ab. Es ist anzumerken, dass ein linearer Zusammenhang zwischen der Elektronentransferdistanz und ln(V) vorliegt (Abbildung 6), was in Übereinstimmung mit einem Superaustauschmechanismus ist.34 Die Steigung der Ausgleichsgeraden liegt bei −0.14±0.01, ein Wert, der vergleichbar mit ähnlich aufgebauten Triaryl- (−0.16)34 und N,N′-Dihydrodimethylphenazinsystemen (DHP; −0.17)33 ist.

Linearer Zusammenhang zwischen ln(V) und der Elektronentransferdistanz (n−1). n gibt die Anzahl der Bindungen zwischen den Stickstoffatomen wieder.
|
R [Å] |
VMH [cm−1] |
λMH [cm−1] |
ΔGǂMH [kJ mol−1] |
g-Wert bei RT |
Linienbreite (FWHM) bei RT [10−4 cm−1/GHz] |
Hyperfeinkonst. A bei RT [10−4 cm−1] |
|---|---|---|---|---|---|---|---|
DTA.+ |
12.5 |
825 |
4147 |
4.5 |
2.0015 |
0.385 |
3.73 |
DTB.+ |
14.3 |
566 |
4489 |
7.5 |
2.0044 |
0.400 |
3.99 |
DTT.+ |
21.2 |
261 |
7147 |
18.4 |
2.0015 |
0.550 |
4.10 |
R133 |
12.5 |
980 |
6600 |
8.9 |
– |
– |
– |
R233 |
12.5 |
870 |
8500 |
10.9 |
– |
– |
– |
Die ermittelte Reorganisationsenergie folgt dem gegensätzlichen Trend. Mit 4147 cm−1 ist diese am kleinsten für DTA.+ und wächst über 4489 cm−1 für DTB.+ zu 7147 cm−1 für DTT.+ stetig an. Eine offensichtliche Erklärung für diesen Trend liegt in der zunehmenden Flexibilität und steigenden Zahl der Rotationsfreiheitsgrade bei längeren Brücken. Ein Vergleich der Stokes-Verschiebung, welche ein Maß für die Reorganisation nach photonischer Anregung darstellt, untermauert diese Interpretation (vergleiche SI Abschnitt 7.1).
Auf der einen Seite fällt die elektronische Kopplung von DTA.+ im direkten Vergleich zu Acetylen-verbrückten Triaryl- und DHP-Dimeren etwas geringer aus. Auf der anderen Seite jedoch ist die Reorganisationsenergie im Vergleich zu diesen nicht-planaren Elektronendonoren erheblich kleiner, wobei DHPs die größte Reorganisationsenergie aufgrund ihrer schmetterlingsartigen Struktur aufweisen.33 Im Vergleich zum Triaryldimer ist die Reorganisationsenergie des DTA.+ um 35 % geringer, was auf die zusätzliche Planarisierung zurückzuführen ist.
 (6)
(6)Daraus lässt sich schlussfolgern, dass eine Minimierung der Reorganisationsenergie auch eine Verringerung der Aktivierungsbarriere zur Folge hat. Für DTA.+ beträgt diese Barriere lediglich 4.5 kJ mol−1. Die Untersuchungen von Lambert et al. am vergleichbaren Triarylderivat R1 ergaben eine doppelt so große Aktivierungsbarriere.33 Dies bekräftigt zusätzlich die positiven Effekte der zusätzlichen Planarisierung in Bezug auf die Elektronentransfereigenschaften der N-HTAs.
Zusammenfassung
Es wurden drei lineare N-HTA-Dimere – DTA, DTB und DTT – mit π-konjugierten Verbindungsstücken verschiedener Längen synthetisiert und ihre Kationen gründlich in Bezug auf gemischtvalenten Ladungstransfer (MV-CT) untersucht. Für die einfach geladenen Radikalkationen wurden mit dem BLYP35-Dichtefunktional lokalisierte elektronische Strukturen vorhergesagt, welche der Robin-Day-Klasse II entsprechen. Bei der chemischen Oxidation konnten für DTA.+, DTB.+ und DTT.+ starke Absorptionsbanden im nIR-Bereich beobachtet werden. Die Analyse dieser Banden im Zusammenhang mit der Mulliken-Hush-Theorie ergab, dass auf der einen Seite die elektronische Kopplung mit der Elektronentransferdistanz abnimmt, wobei auf der anderen Seite die Reorganisationsenergie gleichzeitig zunimmt. Auch diese Analyse legt nahe, dass alle drei MV-Systeme der Robin-Day-Klasse II angehören. Im direkten Vergleich des kleinsten Dimers DTA mit ähnlichen Triaryl- und N,N′-Dihydrodimethylphenazinsystemen ist die Reorganisationsenergie durch die planarisierte Struktur der N-HTAs signifikant reduziert. Letztendlich führt dies sogar zu einer Halbierung der Aktivierungsbarriere im Vergleich zum Triaryldimer. Deswegen stellen planarisierte N-HTAs eine vielversprechende Alternative gegenüber propellerartigen Triarylaminen dar
Acknowledgements
Die Autoren bedanken sich bei der Deutschen Forschungsgemeinschaft (DFG) – Projekt-Nummer 182849149 – SFB 953 und “Solar Technologies Go Hybrid” – eine Initiative der Bayrischen Staatsministeriums für Wissenschaft und Kunst – für die Förderung. Open Access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.
Conflict of interest
Die Autoren erklären, dass keine Interessenkonflikte vorliegen.

