Iridium-Catalyzed Arylative Cyclization of Alkynones by 1,4-Iridium Migration†
We thank the ERC (Starting Grant No. 258580) and the EPSRC (Leadership Fellowship to H.W.L.) for financial support. We thank Dr. Gary S. Nichol (University of Edinburgh) and Dr. William Lewis (University of Nottingham) for X-ray crystallography, and Lorna Eades (University of Edinburgh) for ICP-MS analysis. The EPSRC National Mass Spectrometry Facility is gratefully acknowledged for providing high-resolution mass spectra.
Abstract
1,4-Metal migrations enable the remote functionalization of CH bonds, and have been utilized in a wide variety of valuable synthetic methods. The vast majority of existing examples involve the 1,4-migration of palladium or rhodium. Herein, the stereoselective synthesis of complex polycycles by the iridium-catalyzed arylative cyclization of alkynones with arylboronic acids is described. To our knowledge, these reactions involve the first reported examples of 1,4-iridium migration.
Since the early reports of 1,4-palladium migration1a–1d and 1,4-rhodium migration,2a,2b numerous catalytic reactions involving 1,4-metal migration have been developed.1–5 Such processes enable the remote functionalization of CH bonds, allowing the introduction of metal centers at positions that would otherwise be difficult to metalate. To date, reactions involving the 1,4-migration of palladium,1 rhodium,2 platinum,1q nickel,4 and cobalt5 have been achieved. The demonstration of the ability of other metals to undergo 1,4-migration would be valuable, as their distinct properties may offer new opportunities for the development of useful synthetic methods. Herein, we describe the preparation of highly functionalized polycycles by the iridium-catalyzed arylative cyclization of alkynones. One of the key steps in this transformation is a 1,4-iridium migration, which, to our knowledge, has not been described previously.
During a program aimed at the stereoselective synthesis of complex polycycles by the desymmetrization of cyclic 1,3-diketones,6, 7 we became interested in developing an arylative cyclization of substrates such as 1 a (Scheme 1). We envisaged that in the presence of a suitable metal complex, an arylboron reagent could be employed in an arylmetalation of the alkyne moiety of 1 a to give alkenylmetal species 3. This intermediate could then undergo an alkenyl-to-aryl 1,4-migration to provide intermediate 4, which could then participate in the nucleophilic attack of one of the ketones to give tertiary-alcohol-containing tricycle 2 a.

Proposed arylative cyclization of alkynones.
 (1)
(1) (2)
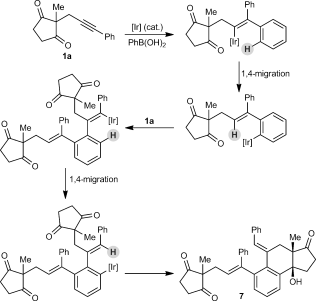
(2)The iridium-catalyzed arylative cyclization of various substrates with PhB(OH)2 was then explored (Scheme 2). In all reactions, the 2:1 adduct was observed in approximately 10–25 % yield by 1H NMR analysis of the reaction mixtures, but these products were not isolated. Substituents at the para, meta, or ortho positions of the aryl group on the alkyne were tolerated (2 b–f), though in the case where an ortho-cyano group was present, a higher loading of [{Ir(cod)Cl}2] (2.5 mol %) was required for full conversion (2 f). With para-substituted phenyl groups, electron-poor rather than electron-rich arenes led to higher yields of the products (compare 2 b–d), which is likely due to a more regioselective initial arylmetalation of more polarized alkynes. The relative configurations of the stereogenic centers and the E geometry of the alkenes in the products were assigned by analogy with 2 d, the structure of which was determined by X-ray crystallography.9 Substrates containing a terminal alkyne or an alkyne lacking an aryl substituent did not undergo the reaction and returned only unreacted starting material (2 g and 2 h).

Arylative cyclization of various alkynones. Reactions were conducted using 0.40 mmol of 1 a–n in toluene (4 mL). Cited yields are of isolated products. [a] Compound 7 was also isolated in 27 % yield. See Equation (2). [b] 2.5 mol % of [{Ir(cod)Cl}2] was used.
Next, variations of the pendant ketone were examined. An indane-1,3-dione reacted well to give 2 i in 67 % yield. Changing the substituent at C2 (between the ketones) from a methyl to a phenyl group was tolerated, and 2 j was obtained in 63 % yield using 2.5 mol % of [{Ir(cod)Cl}2]. Switching from five- to six-membered ring diketones was also possible (2 k–m9). In these cases, and in a similar fashion to the five-membered ring substrates, the reactions of substrates containing more electron-deficient arenes on the alkyne led to higher yields than those with electron-rich arenes (compare 2 k–m). A cyclic β-ketoamide was also tolerated, providing 2 n in 72 % yield.
 (3)
(3)Table 1 presents the results of arylative cyclization of 1 a with various arylboronic acids. The reaction was compatible with methyl (Table 1, entry 5), methoxy (Table 1, entry 1), halide (Table 1, entries 1 and 5), or ester groups (Table 1, entry 3) at either the para or meta positions of the arylboronic acid. However, with electron-withdrawing substituents, a higher catalyst loading (5 mol % of Ir) was required for acceptable yields (Table 1, entries 2, 3, and 5). With a 4-carboethoxy group, the yield was lower (35 %), and unreacted 1 a was recovered in 41 % yield (Table 1, entry 3). 2-Naphthylboronic acid also reacted smoothly to give 10 f in 59 % yield (Table 1, entry 6). Importantly, the reactions of meta-substituted arylboronic acids were highly regioselective (≥10:1 regioisomeric ratio, determined by 1H NMR analysis of the unpurified reaction mixtures) and provided 10 d–f as the major products (Table 1, entries 4–6). These results demonstrate that there is a strong preference for iridium to undergo 1,4-migration to the sterically least hindered site of the arene.12

|
Entry |
Ar |
Product |
Yield [%][b] |
|
|---|---|---|---|---|
|
1 2 3 |
4-MeOC6H4 4-ClC6H4 4-EtO2CC6H4 |
|
10 a R=OMe 10 b R=Cl 10 c R=CO2Et |
69 62[c] 35[c,d,e] |
|
4 5 |
3-MeC6H4 3-BrC6H4 |
|
10 d R=Me 10 e R=Br |
68[f] 58[c,f] |
|
6 |
2-naphthyl |
|
10 f |
59[c,f,g] |



- [a] Reactions were conducted with 0.40 mmol of 1 a in toluene (4 mL). [b] Yields of isolated products. [c] 2.5 mol % of [{Ir(cod)Cl}2] was used. [d] 3.0 equiv each of ArB(OH)2, KF, and tBuOH were used. [e] Substrate 1 a was recovered in 41 % yield. [f] Single regioisomer observed. [g] Reaction conducted at 90 °C.
 (4)
(4)A possible catalytic cycle for these transformations, using 1 a and PhB(OH)2 for illustrative purposes, is shown in Scheme 3. First, an aryliridium species 12 is generated by transmetalation from the arylboronic acid to the iridium butoxide 11 (or alternatively, an iridium fluoride). Migratory insertion of the alkyne into 12 then occurs to give alkenyliridium species 13,13, 14 which then undergoes 1,4-migration. The resulting aryliridium intermediate 14 then undergoes nucleophilic attack onto one of the ketones to give iridium alkoxide 15. Protonation of 15 with tBuOH releases the product 2 a and regenerates the iridium butoxide 11.

Proposed catalytic cycle for the arylative cyclization.
 (5)
(5) (6)
(6)In summary, we have reported the iridium-catalyzed arylative cyclization of alkynones with arylboronic acids.16 These reactions involve 1,4-iridium migration as a key step, a mode of reactivity for iridium that, to our knowledge, has not been reported previously.17 Efforts to exploit the 1,4-migration of iridium and other metals in new catalytic transformations are ongoing in our group.