

CSF3R T618I Collaborates With RUNX1-RUNX1T1 to Expand Hematopoietic Progenitors and Sensitizes to GLI Inhibition
Supplemental digital content is available for this article.
Graphical Abstract

Abstract
Activating colony-stimulating factor-3 receptor gene (CSF3R) mutations are recurrent in acute myeloid leukemia (AML) with t(8;21) translocation. However, the nature of oncogenic collaboration between alterations of CSF3R and the t(8;21) associated RUNX1-RUNX1T1 fusion remains unclear. In CD34+ hematopoietic stem and progenitor cells from healthy donors, double oncogene expression led to a clonal advantage, increased self-renewal potential, and blast-like morphology and distinct immunophenotype. Gene expression profiling revealed hedgehog signaling as a potential mechanism, with upregulation of GLI2 constituting a putative pharmacological target. Both primary hematopoietic cells and the t(8;21) positive AML cell line SKNO-1 showed increased sensitivity to the GLI inhibitor GANT61 when expressing CSF3R T618I. Our findings suggest that during leukemogenesis, the RUNX1-RUNXT1 fusion and CSF3R mutation act in a synergistic manner to alter hedgehog signaling, which can be exploited therapeutically.
INTRODUCTION
Somatic mutations of the colony-stimulating factor-3 receptor (CSF3R) gene in acute myeloid leukemia (AML) are highly associated with alterations of hematopoietic transcription factors, such as core binding factor (CBF) abnormalities or CEBPA double mutations.1-3 In particular, we and others found proximal membrane domain mutations of CSF3R, represented by the hotspot alteration T618I, either in t(8;21) positive CBF leukemia or CEBPA double mutated AML.4-6 CSF3R T618I was initially described in chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML).4,7 Proximal membrane mutations of CSF3R lead to ligand independent receptor activation with disrupted differentiation and proliferative advantage. While the presence of the t(8;21) associated fusion gene RUNX1-RUNX1T1 in AML generally indicates a favorable prognosis, we lack understanding of the role of CSF3R mutations during AML development and treatment outcome. RUNX1-RUNX1T1 (also known as AML1-ETO) leads to partial block of myeloid differentiation, but is not sufficient to cause leukemia alone, indicating the requirement of additional genetic lesions.8-11 The cooperation between alterations in hematopoietic transcription factors and signaling cascades leads to block of differentiation and increased proliferation, which represents a classical model of leukemogenesis.12,13 In the present study, we investigated the oncogenic collaboration between the RUNX1-RUNX1T1 fusion and the CSF3R mutant T618I and found that—beyond disturbing differentiation and driving proliferation in a complementary manner—they may synergistically activate specific pathways.
METHODS
Plasmids
The expression vector pMSCV-RUNX1-RUNX1T1TR-IRES-tdTomato has been described previously.14 Expression vectors pMSCV-IRES-GFP with CSF3R wild type and T618I mutant, and vector control were a generous gift from Julia Maxson, Portland, Oregon.15
Cell culture and retroviral transduction
SKNO-1 cells (DSMZ, ACC-690) were cultured in RPMI 1640 Glutamax (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (PAN Biotech, Aidenbach, Germany), 1% penicillin-streptomycin (PAN biotech), and 10 ng/mL granulocyte-macrophage colony stimulating factor (GM-CSF) (Miltenyi Biotech, Bergisch Gladbach, Germany). HEK 293T cells (DSMZ, ACC-635) were cultured in Dulbecco’s modified Eagle’s medium (PAN Biotech) supplemented with 10% FBS and 1% penicillin-streptomycin. CD34+ bone marrow cells (Lonza, Basel, Switzerland) were cultured in Iscove’s modified Dulbecco’s medium (Gibco, Carlsbad, CA), supplemented with 20% FBS, 1% penicillin-streptomycin, 4 µM L-Glutamine (Gibco), 10 ng/mL interleukin (IL)-3, 20 ng/mL IL-6, 20 ng/mL Flt3-L, 20 ng/mL GM-CSF, 20 ng/mL stem cell factor, and 20 ng/mL thrombopoietin (cytokines were obtained from Peprotech, Hamburg, Germany). Retroviral transduction of CD34+ cells was performed as described before.14,16 Transduction of SKNO-1 was performed accordingly with the culture medium described above.
Flow cytometry
Beginning 4 days posttransduction, CD34+ progenitor cells were analyzed every 2–3 days for expression of tdTomato and enhanced green fluorescent protein (GFP) by flow cytometry. SKNO-1 cells were either sorted 10 days after transduction or left unsorted.
For analysis of cell surface marker expression, allophycocyanin (APC)-, phycoerythrin/cyanine 7 (PE/Cy7)-, or PE-Vio770-conjugated anti-human HLA-DR, CD3, CD10, CD11b, CD11c, CD13, CD14, CD15, CD18, CD19, CD27, CD33, CD34, CD41a, CD49a, CD117, and CD135 antibodies were used (BioLegend, San Diego, CA; BD Life Sciences, Franklin Lakes, NJ; or Miltenyi Biotech, Bergisch Gladbach, Germany). Isotype controls mouse IgG1, mouse IgG2a, mouse IgM, and recombinant human IgG1 were used accordingly. CSF3R localization was evaluated in permeabilized SKNO-1 cells using PE-conjugated (intracellular) or APC-conjugated (surface) anti-human CD114 antibody (Miltenyi Biotech, Bergisch Gladbach, Germany). Mouse IgG1, k isotype controls for PE and APC (BD Biosciences, Franklin Lakes, NJ) were used as control.
Apoptosis rates were measured by flow cytometry following cell staining with APC-conjugated Annexin V (BD Pharmingen, Franklin Lakes, NJ) according to the manufacturer’s recommendations in combination with 4',6-diamidino-2-phenylindole (DAPI) as vital dye.
Cell cycle was assessed by flow cytometry after DRAQ5 staining (Alexis Biochemicals, San Diego, CA) for 10 minutes at 37°C.
All flow cytometry experiments were performed using FACS Calibur or FACS Canto II (BD Life Sciences). Data were analyzed using FlowJo Software (BD Life Sciences). Statistical analysis was performed using GraphPad Prism (GraphPad Software Inc., San Diego, CA). P-values were calculated using the student t test.
Cytotoxicity assay
SKNO-1 or progenitor cells (3 × 104/well) were treated in 96-well plates for 72 hours with varying concentrations of Janus kinase (JAK) inhibitor ruxolitinib phosphate (ChemScene, Monmoth Junction, NJ), STAT3 inhibitor XIII, C188-9 (EMD Millipore, Billerica, MA), GLI inhibitor GANT61 (TOCRIS, Bristol, UK), or the multikinase (BCR-ABL/SRC) inhibitor dasatinib (Santa Cruz Biotechnology, Dallas, TX). Sixty-eight hours posttreatment, 20 µL of CellTiter-Blue reagent was added to each well of cells as recommended by the manufacturer. Plates were incubated for additional 4 hours at 37°C in the dark. Readout was obtained using the CellTiter-Blue Cell Viability Assay protocol with the GloMax Discover plate reader (both from Promega, Fitchburg, WI). Three independent experiments, each consisting of 3 technical replicates for the single-drug concentrations, were performed for the inhibitors tested. Analysis was carried out in GraphPad Prism (GraphPad Software Inc., San Diego, CA). Values were normalized to the dimethyl sulfoxide control (drug solvent) and drug response was assessed by nonlinear regression (curve fit) using the equation: “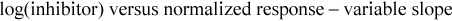 ”.
”.
Bulk and single-cell RNA sequencing
Human progenitor cells double transduced with RUNX1-RUNX1T1tr and CSF3R WT or T618I were submitted to bulk RNA sequencing (Prime-seq) at day 60 of outgrowth. Library preparation, sequencing, analysis of differential gene expression, and pathway analysis were performed as described before.16-18 For single-cell RNA sequencing (scRNA-seq), libraries from cells harvested on day 30 and 60 were prepared using the Chromium Next GEM Single Cell 3′ GEM Kit v3.1 (10× Genomics, Pleasanton, CA) according to the manufacturer’s instructions. Paired-end sequencing (Read 1: 28bp; Read 2: 91bp) was performed on an Illumina HiSeq 1500 instrument, with an average of 50,000 reads per cell and 10,000 cells per library. Sample demultiplexing and alignment of the data were done using the Cell Ranger software (v6.1.2) and the human genome GRCh38.p13 from Gencode (release 39). Further data processing (quality control, filtering, normalization, highly variable gene selection, embedding, and visualization) was performed using the python package scanpy v1.9.1.19 We filtered cell barcodes with <3000 counts, >100,000 counts, or >20% of mitochondrial reads. Additionally, we removed cells with >600 genes captured. It resulted in 29,781 cells and 30,039 genes across 4 samples (at day 30: 7530 cells for CSF3R WT and 7661 cells for CSF3R T618I; at day 60: 6432 cells for CSF3R WT and 8158 cells for CSF3R T618I, respectively). Finally, the cell type annotation was done using the package CellTypist v1.3.1.20 The notebooks used for the scRNA-seq analysis are available on Github (https://github.com/colomemaria/CSF3R_scRNAseq_reproducibility).
RESULTS
CSF3R T618I confers a clonal advantage to RUNX1-RUNX1T1 expressing progenitors
We set out to study the role of the activating CSF3R mutation T618I in a RUNX1-RUNX1T1 positive background, using CD34+ hematopoietic stem and progenitor cells (HSPCs) from adult healthy donors (Figure 1A). While expression of a truncated form of the t(8;21) related RUNX1-RUNX1T1 (RUNX1-RUNX1T1tr) fusion is sufficient to expand hematopoietic progenitor cells, we observed that coexpression of the CSF3R mutant T618I (CSF3R T618I) but not CSF3R wild type (WT) led to competitive outgrowth of cells (Figure 1B; Suppl. Figure S1A-S1C). In contrast, overexpression of either CSF3R WT or CSF3R T618I alone did not lead to considerable cell expansion (Suppl. Figure S1D). The CSF3R single transduced hematopoietic progenitor cells started to differentiate and lost their proliferative potential within 1 month (data not shown).
CSF3R T618I induces an immunophenotype resembling acute myelomonocytic leukemia
After ex vivo expansion, cells were analyzed via flow cytometry for surface expression of differentiation markers. Only a minor subpopulation of RUNX1-RUNX1T1tr single positive cells showed surface expression of CD34, while most cells expressed myeloid markers, consistent with previous reports.11,14,21 CSF3R T618I coexpressing cells were negative for CD34, but highly positive for the pan-myeloid marker CD13 and the monocytic marker CD14. In addition, double positive cells expressed HLA-DR, CD11b, CD11c, CD33, CD18, and partly CD49a (Suppl. Figure S2). A similar immunophenotype was reported in monocytic cells, including monoblasts and promonocytes, of patients with acute myelomonocytic leukemia (AMML).22-24

CSF3R T618I collaborates with RUNX1-RUNX1T1tr to expand HSPCs. (A) Schematic outline of the experimental setup. hCD34+ cells were virally transduced with CSF3R WT or T618I (GFP) and RUNX1-RUNX1T1tr (tdTomato). Competitive growth was assessed by flow cytometry every 2–3 d over a period of 60 d. (B) Flow cytometry measurement of one representative competitive growth assay. (C) Limiting dilution assay: indicated cell numbers of transduced hCD34+ cells were seeded after expansion. Cell number was assessed 11 d post seeding (left). Serial dilution: 1 × 105 transduced hCD34+ cells were seeded after expansion. Cells were counted every 2–3 d and reseeded at initial cell number (right). Error bars represent ±SEM. CSF3R = colony-stimulating factor-3 receptor; HSPC = hematopoietic stem and progenitor cells; WT = wild type.
CSF3R T618I leads to increased self-renewal and blast morphology
Because self-renewal potential and increased proliferation frequency are key mechanisms in leukemogenesis, we performed limiting and serial dilution experiments. Seeding low numbers of RUNX1-RUNX1T1tr single positive cells did not result in measurable proliferation after 7 or 11 days, whereas seeding of 1 × 104 cells resulted in an increase of cell number by 50% after 11 days. Analysis of double oncogene expressing cells 11 days post seeding revealed an average increase of cell number by 15-fold to 30-fold (Figure 1C, left panel). Serial dilution to 1 × 105 cells showed highly increased proliferation and self-renewal potential in double oncogene expressing cells as compared with RUNX1-RUNX1T1tr single positive cells (Figure 1C, right panel).
To mimic the subsequent acquisition of genetic lesions required for leukemic transformation, we next transduced naive HSPCs with RUNX1-RUNX1T1tr alone. Thereby, the cells clonally expanded, resulting in a culture composed of >95% fluorescence-marker positive cells. Next, we introduced a second hit by superinfection with the retroviral CSF3R T618I construct (Figure 2A). This led to complete outgrowth of double positive cells over a period of another 60 days (Figure 2B). The cytomorphology analysis showed a heterogeneous mixture of mature and immature cells in RUNX1-RUNX1T1tr single positive cells, whereas double positive cells remained mostly undifferentiated, featuring a homogeneous blast-like morphology (Figure 2C).
CSF3R T618I and RUNX1-RUNX1T1 synergistically alter specific pathways
Next, we analyzed the differential gene expression between single and double oncogene expressing primary HSPCs upon outgrowth (day 60, n = 6/group). RNA sequencing by Prime-seq18 revealed a significant deregulation of 626 genes upon expression of CSF3R T618I (Figure 3A; Suppl. Table S1). Pathway analysis showed alteration of folate and pterin metabolism, a central mechanism of nucleotide synthesis and DNA methylation. Furthermore, signaling pathways of IL-10, IL-4, and IL-13 were upregulated, all known to have anti-inflammatory properties on immune cells. Interestingly, also the hedgehog signaling pathway was significantly enriched (Figure 3B) with GLI2 being among the most prominently upregulated genes upon CSF3R T618I expression (Figure 3A and 3C). Consistent with previous reports,25 we observed shorter overall survival in patient samples of the German Acute Myeloid Leukemia Cooperative Group (AMLCG)-2008 trial (Figure 3D). In gene expression data from Beat-AML patients, we confirmed upregulation of GLI2 in patients harboring FLT3-ITD (Suppl. Figure S3A). Of note, within the Beat-AML cohort, we found elevated levels of GLI2 expression in AML patients with CSF3R mutations, similar to AML with FLT3-ITD (Suppl. Figure S3B; Suppl. Table S2).

Forced expression of CSF3R T618I in a RUNX1-RUNX1T1tr background provides a competitive advantage and induces blast morphology. (A) Schematic outline of the experimental setup. hCD34+ cells were virally transduced with RUNX1-RUNX1T1tr and expanded over the period of 60 d. Expansion of tdTomato-positive cells was assessed every 2–3 d by flow cytometry. Post expansion, hCD34+ cells were superinfected with CSF3R WT or T618I. Competitive growth was assessed by flow cytometry every 2–3 d over a period of another 60 d. (B) Flow cytometry measurement of one representative competitive growth assay after superinfection. (C) Morphological analyses of cytospin/Giemsa preparations of hCD34+ cells 60 d post superinfection. CSF3R = colony-stimulating factor-3 receptor; WT = wild type.
We next performed single-cell RNA-seq in order to evaluate the dynamic changes in the double transduced cell populations over time (Suppl. Figure S4). Upon outgrowth (day 60), a representative culture with CSF3R T618I-mediated expansion of double positive cells showed more immature hematopoietic stem and multipotent progenitor cells (HSC/MPP) compared with the culture with RUNX1-RUNX1T1tr-mediated expansion of single positive cells and cotransduction with CSF3R WT (Figure 4A; Suppl. Figure S4b). This is in line with the cytomorphology featuring predominantly blast-like cells in the double positive culture (Figure 2C). Furthermore, GLI2 expression was enriched in the HSC/MPP compartment and in the macrophage-committed progenitors, the two most abundant cell types within the CSF3R T618I transduced culture (Figure 4B and 4C).

CSF3R T618I induces Hedgehog signaling. (A) Heat map of significantly (P < 0.05) deregulated genes in hCD34+ cells after 60 d of competitive outgrowth with RUNX1-RUNX1T1tr and CSF3R T618I compared with cotransduction of vector control (EV) or CSF3R WT. Data was obtained after performance of bulk RNA-seq (Prime-Seq) analysis. (n = 2 independent transductions/construct; n = 6 replicates/transduction). (B) GSEA of RNA-seq data shows enrichment of hedgehog signaling genes in CSF3R T618I expressing cells. (C) GLI2 expression as obtained from RNA-seq. Replicates of outgrown RUNX1-RUNX1T1tr populations (in red) resulting from double transduction with CSF3R WT (n = 12) and pMIG EV (n = 11) were pooled together for this analysis, as they behave similarly in the growth competition assay. Differential expression analysis was performed with edgeR/limma package (****P < 0.0001). (D) OS according to GLI2 expression of patients enrolled in the AMLCG-2008 study cohort (GSE106291). High expression was considered n(Transcripts) ≥1; low/no expression was considered n(Transcripts) <1. CSF3R = colony-stimulating factor-3 receptor; GSEA = gene set enrichment analysis; OS = overall survival; WT = wild type.
CSF3R T618I confers cytokine independence in the t(8;21)-positive cell line SKNO-1
Previous studies in murine t(8;21) models have shown that preleukemic clones, expanded by RUNX1-RUNX1T1, are highly cytokine dependent, suggesting that mutations leading to cytokine-mediated survival and proliferation advantages might be crucial collaborators in leukemogenesis.11,21,26,27 Therefore, we conducted further experiments in the GM-CSF dependent t(8;21)-positive cell line SKNO-1, where we observed cytokine independent growth in the presence of CSF3R T618I but not CSF3R WT (Figure 5A). GM-CSF withdrawal of SKNO-1 native cells, vector control, or CSF3R WT expressing cells led to cell cycle arrest that could be overcome by expression of CSF3R T618I (Figure 5B). Because activation of the receptor by its ligand G-CSF physiologically requires dimerization and internalization, we sought to characterize surface and intracellular abundance of CSF3R. SKNO-1 cells overexpressing CSF3R WT showed higher levels of receptor protein on the surface. In contrast, overexpressed CSF3R T618I localized predominantly in the intracellular compartment, while the ratio of surface to intracellular localization did not differ markedly from endogenous CSF3R (Suppl. Figure S5).

Single-cell RNA-seq shows enrichment of immature hematopoietic cells (HSC/MPP) and GLI2 upregulation in the culture with CSF3R T618I-mediated expansion of double positive cells (day 60). (A) UMAP showing different subpopulations annotated with CellTypist for CSF3R WT or T618I. (B) UMAP showing GLI2 expression for CSF3R WT or T618I. (C) Violin plots showing GLI2 expression across the subpopulations for CSF3R WT or T618I. CSF3R = colony-stimulating factor-3 receptor; HSC/MPP = hematopoietic stem and multipotent progenitor cells; WT = wild type.
CSF3R T618I sensitizes to GLI inhibition in a RUNX1-RUNX1T1 background
Subsequently, we investigated pharmacological counteraction of CSF3R T618I through inhibition of putative downstream effectors. Therefore, we used GANT61 to treat outgrown HSPCs that express RUNX1-RUNX1T1tr in combination with either CSF3R WT or CSF3R T618I. Expression of CSF3R T618I significantly sensitized the cells to GLI inhibition (Figure 5C; right panel).

CSF3R T618I confers cytokine independence and increased sensitivity to GLI inhibition. (A) Competitive growth analysis of cell line SKNO-1 virally transduced with vector control (EV), CSF3R WT or T618I, cultivated with or without GM-CSF. Expression of fluorescent marker (GFP) was assessed every 2–3 d using flow cytometry. (B) Cell cycle analysis of SKNO-1 untransduced, transduced with vector control (EV), CSF3R WT or T618I, and cultured with or without GM-CSF. Cell cycle was assessed after staining with DRAQ5 and detection using flow cytometry. Cell cycle analysis was performed using FlowJo Software. Error bars represent ±SEM. Comparison was performed using two-tailed unpaired t test (****P < 0.0001). (C) GLI inhibition using escalating doses of GANT61 in sorted SKNO-1 (left) and expanded hCD34+ cells (right) transduced with CSF3R WT or T618I. CSF3R = colony-stimulating factor-3 receptor; GM-CSF = granulocyte-macrophage colony stimulating factor; WT = wild type.
Independently, we confirmed that CSF3R T618I expressing SKNO-1 cells were as well significantly more sensitive toward treatment with GANT61, as compared with CSF3R WT control (Figure 5C; left panel). Considering previously reported signal transduction of mutated CSF3R via JAK2 and STAT3,7 we also tested the JAK2 inhibitor ruxolitinib and the STAT3 inhibitor C188-9. Pharmacological targeting of either of the two downstream effectors did not show increased sensitivity upon ectopic expression of CSF3R T618I compared with WT (Suppl. Figure S6A and S6B). Remarkably, Western Blot analysis showed enhanced phosphorylation of STAT3 for CSF3R T618I as compared with CSF3R WT or vector control, whereas JAK2 protein expression was highly decreased with no measurable phosphorylation (Suppl. Figure S6D). Interestingly, CSF3R T618I conferred a slightly increased sensitivity to the multikinase inhibitor dasatinib (Suppl. Figure S6C).
DISCUSSION
Although the CSF3R mutation T618I has been investigated extensively in the context of CNL and aCML,7,28-30 its role in AML is mainly described in conjunction with CEBPA mutation.31 In a first step, we were able to shed light on the importance of the CSF3R mutation T618I in clonal competition during leukemogenesis. It is widely believed that the RUNX1-RUNX1T1 fusion leads to enhanced self-renewal potential and blockade of various differentiation pathways, thus expanding early progenitor cells.8,10,11,21,32 Markedly, hematopoietic progenitor cells retrovirally expressing RUNX1-RUNX1T1 reportedly do not promote leukemia in immunodeficient mice.8-11 While treatment with G-CSF has been shown to increase cell proliferation significantly upon expression of RUNX1-RUNX1T1,33 leukemogenesis only occurs in the presence of secondary mutations.8,10,27,34-37 According to the classical two-hit model, leukemogenesis in AML requires a block of differentiation on the one hand and increased proliferative advantage on the other.12 In accordance with the literature, we were able to expand hematopoietic stem and progenitor cells by ectopically expressing RUNX1-RUNX1T1tr, leading to blockage of differentiation. Interestingly, CSF3R T618I not only enhanced proliferation and provided cytokine independent cell cycle progression but also generated immature cell populations with even higher frequency of cells with self-renewal potential and increased survival potential. Thus, CSF3R T618I does not only serve as a classical proliferation driver in our setting, but also further aggravates the block of differentiation. Coexpression of RUNX1-RUNX1T1tr and CSF3R T618I highly resembled the phenotype of AMML, all together suggesting leukemic transformation.
CSF3R signaling has been proposed to involve phosphorylation of JAK2 and STAT3 in the context of CNL.7 Furthermore, activating JAK2 mutations are recurrently found in t(8;21) positive AML, suggesting a cooperative effect in leukemogenesis.5,38-41 Interestingly, pharmacologic counteraction of both JAK2 and STAT3 signaling did not result in increased cell death upon expression of CSF3R T618I in our study. Here, we found a significant decrease in JAK2 protein abundance (Suppl. Figure S6D) that was not reflected in significant mRNA deregulation (Suppl. Figure S7A). This suggests both a posttranslational downregulation mechanism of JAK2, possibly by SOCS342,43 (Suppl. Figure S7C), and an alternative activation of STAT3 in presence of mutated CSF3R. The increased phosphorylation of STAT3 upon CSF3R T618I expression (Suppl. Figure S6D) points toward an alternative, possibly redundant signal transduction of mutated CSF3R in the context of t(8;21) AML. Alternative pathways might involve signaling via ERK15,44 or a switch toward different members within the JAK family. This is supported by our RNA-seq results, where we found JAK3 significantly upregulated upon CSF3R T618I expression (Suppl. Figure S7B).
Remarkably, CSF3R T618I SKNO-1 cells were more sensitive to dasatinib, which is classically defined as a BCR-ABL and SRC family kinase inhibitor. STAT3 is one of the downstream targets of SRC signaling pathway; however, as STAT3 inhibition did not render the CSF3R T618I expressing cells more sensitive, signaling through the SRC kinases may involve other downstream effectors.45
As an alternative route, we observed that mutated CSF3R induces ligand independent signaling via the hedgehog pathway. In particular, we found the hedgehog downstream effector GLI2 specifically upregulated upon coexpression of RUNX1-RUNX1T1 and CSF3R T618I. Of note, patient data and mouse models support a specific collaboration of upregulated GLI2 and FLT3-ITD in AML.25,46 GLI2 expression is a negative prognostic marker in AML, associated with significantly shorter event-free and overall survival,25 as we were able to confirm within the AMLCG patient cohort. The correlation between GLI2 expression and overall survival is not limited to the CBF subgroup, suggesting a general mechanism in AML. Hedgehog pathway signaling plays a critical role in embryogenesis, primitive embryonal hematopoiesis, and maintenance of hematopoietic stem cells and is tightly regulated.47 Deregulation in AML might lead to development and expansion of chemotherapy resistant leukemic stem cells, promoting therapy resistance and relapse.48,49 Thus, inhibition of hedgehog pathway signaling became a promising target. Recently, the smoothened (SMO) inhibitor glasdegib was approved both by the United States Food and Drug Administration (US FDA) and the European Medicines Agency (EMA) as the first hedgehog pathway inhibitor for treatment of AML patients in combination with low-dose cytarabine (Ara-C),50,51 while further drugs are investigated. In preclinical studies, GLI inhibition by GANT61 caused apoptosis in Ara-C-resistant cells and sensitized to both Ara-C and rapamycin treatment.48,52,53 Interestingly, targeting of the hedgehog pathway sensitized to dasatinib treatment in pancreatic cancer54 and in leukemia cells.55 In light of our results that CSF3R T618I sensitizes both to dasatinib and GANT61, it is tempting to speculate that dasatinib may also target hedgehog signaling components, especially taking into account the reported crosstalk between hedgehog and the SRC downstream effector PI3K/AKT.56 Maxson and colleagues (2013) found that patient-derived cells with CSF3R T618I were resistant to dasatinib. Although the KIT N822K mutation in the SKNO-1 cells might be a potential confounder in our study, since it is reported that this mutation can render these cells more sensitive to dasatinib,57 the increased sensitivity upon expression of CSF3R T618I to this drug is specific. Several studies reported a broad target profile and recognized that dasatinib’s antitumor activity is linked to its promiscuous nature in different cancers.58-61 Therefore, the interplay between hedgehog signaling and potential context-specific targets of dasatinib needs further investigation.
Taken together, we have developed a human in vitro model for clonal competition mimicking the onset and progression of leukemic transformation. Our experiments provide evidence for the oncogenic collaboration between RUNX1-RUNX1T1 and CSF3R T618I via increased hedgehog signaling, which could be counteracted by GLI inhibition. This novel mechanism was confirmed in two independent model systems, namely the cell line SKNO-1 and primary hematopoietic stem and progenitor cells. Considering its upregulation in FLT3-ITD positive AML and upon presence of pathogenic CSF3R mutations, the downstream effector GLI2 becomes an increasingly attractive target for pharmacological intervention in AML.
AUTHOR CONTRIBUTIONS
ASS, LC-W, WE, CW, MC, and PAG designed the study. ASS, VCA, RW, ERM, TH, SK, HB, and MS performed research. ASS, RW, JWB, and AC collected data. ASS, VCA, PAG, and CW interpreted data. AD and PK performed bioinformatics analysis. JWB and MC-T provided bioinformatics support. AS performed statistical analysis. ASS, VCA, and PAG wrote the article.
DATA AVAILABILITY
The transcript expression data from this study are available in the Gene Expression Omnibus (GEO) repository, accession numbers GSE230755 (bulk Prime-seq) and GSE232187 (single-cell RNA-seq).
DISCLOSURES
The authors have no conflicts of interest to disclose.
SOURCES OF FUNDING
This study was supported by the German Research Foundation (DFG) within the Collaborative Research Centre (SFB) 1243 “Cancer Evolution” (Projects A08, A14, and Z02). PAG and CW acknowledge support by the Wilhelm Sander-Stiftung (Förderantrag Nr. 2014.162.3). PAG received funds from the Munich Clinician Scientist Program (MCSP) Advanced Track. MC-T and AD acknowledge the “Initiative and Networking Fund” of the Helmholtz Association (grant VH-NG-1219 for MC-T). AD received funds of the German Research Foundation (DFG STR 1385/5-1).

