

Synthesis and biological activity of an Anderson polyoxometalate bis-functionalized with a Bombesin-analog peptide
Abstract
Polyoxometalates (POMs) are multi metallic and polyanionic oxides whose functionalization with organic moieties can generate new properties. Among possible strategies to generate molecular diversity and find applications in different fields, the postfunctionalization of a POM with peptides is particularly interesting and easily achievable. In this article, we present the functionalization of the Anderson-Evans polyoxomolybdate ([MnMo6O24]3–) with a Bombesin antagonist peptide, to highlight the interplay between these 2 domains, in terms of structural changes and assembly. Moreover, since Bombesin analogs show a marked binding affinity and specificity for some subtypes of the Gastrin Releasing Peptide Receptor (GRP-R), the impact of the peptide on the antitumor activity of the Anderson-Evans polyoxomolybdate POM has been explored.
Graphical Abstract

1 INTRODUCTION
Among small inorganic systems with biological activity, polyoxometalates (POMs) are multimetallic and polyanionic oxides, with interesting potential applications in nanomedicine as low cost antibacterial,1-4 antiviral,5-7 and antitumoral8-10 agents.
Despite their negative charge, several proof-of-concept studies showed that POMs can cross cell membranes, with11-18 or without the involvement of carrier systems, to be mainly localized in the cytoplasm.19-22 Their biological activity mainly stems from the redox behavior,23 which makes them excellent electron acceptors, and from the capability to interact with biological macromolecules through electrostatic interactions. In particular, their affinity towards the cationic pockets of proteins24, 25 and enzymes26, 27 is remarkable, owing to their nanosized dimensions, rigidity, and high charge density. This interaction may lead to misfolding, aggregation or denaturation of peptides and proteins,3, 24, 28 competition with natural, negatively charged, substrates25, 26, 29, 30 or even to proteolytic reactions.31, 32
Hybridization of such inorganic compounds upon organic derivatization with organic/organometallic moieties can generate new properties, springing form a synergistic effect between the 2 domains.33-35 POMs with organic pendants also offer novel opportunities for the development of POM-based nanodrugs,5, 36-43 whereby the organic decoration often represents a valuable tool to increase their stability under physiological pH conditions, while increasing the biocompatibility and reducing the cytotoxicity. The preparation of POM-based surfactants, incorporating lipophilic pendant chains, for example, has recently shown to enhance cell internalization, thanks to the formation of POM-based vesicles with improved selectivity against cancer cells.44 Moreover, a great potential to impart recognition capabilities upon integration of targeting moieties,45 or to enable fluorescence imaging by adding suitable luminescent tags46 is envisaged.
Anderson-Evans polyoxoanions with formula [XM6O24]n– are made of 6 octahedric MO6 (M = Mo or W) which share one of their edges, and with a further templating heteroatom-centered octahedric unit (XO6).47 The resulting planar structure has an approximate D3d symmetry.48 Three different coordination modes of the oxygen atoms can be recognized: 6 μ3-O oxygen atoms (shared by the heteroatom X and 2 M), 6 μ2-O oxygen atoms between 2 M ions, and 12 terminal oxygens, occurring as couples of Ot on each M ion.49 Average dimensions of this compounds are 8.6 × 8.6 × 2.7 Å and their properties (magnetic, redox, luminescence) can be changed depending on heteroatom, type of counter ions, organic functionalization, and resulting symmetry. Being hydrolytically and oxidatively stable, these POMs are suitable for biological applications. The derivatization of Anderson POM with organic molecules can be accomplished by reaction with tris(hydroxymethyl)aminomethane (TRIS), or its (RC(CH2OH)3) analogs with R= -OH, -CH2OH, condensed to the POM through the oxygen atoms μ3-O or μ2-O.49 Depending on the heteroatom, indeed, a different connectivity is accessible, and include both single-sided and double-sided functionalization, as well as asymmetric decoration with different moieties.50, 51
The POM [MnMo6O18((OCH2)3CNH2)2]3– has a recognized activity against cancer.52 Its derivatization can be obtained by means of prefunctionalization or postfunctionalization strategies, which exploit the formation of amide, imide or ester bonds, before or after the conjugation of the tripodal ligand with the POM.47 Some examples of bio-conjugates have been described by Wang and coworkers,53 which have used cholesterol, dehydrocholic, and cholic acid as targeting units for farnesoid X receptor (FXR), a nuclear bile acid receptor, on breast cancer cells. The molybdate bis-functionalyzed with cholic acid, in particular, displays a median inhibitory concentration (IC50) ranging from 38 to 56 μM, which represents a noteworthy improvement with respect to the choline-free POM precursor (IC50 = 200–300 μM), and shows a much lower antiproliferative effect on noncancerous breast epithelial cells (IC50 = 280 μM).
Among the available targeting biomolecules, such as monoclonal antibodies, aptamers, and receptor-specific substrates, peptides are flexible molecules which can display high affinity toward a receptor and impart a high cell penetrating capability, with low immunogenicity.54 Peptides are suitable pendants for POMs55 and can be covalently grafted via amide bonds.56-59 On the other hand, as observed for the targeted proteins mentioned above, they can also establish multiple weak interactions with the POM surface, being these usually more efficient upon increasing of size and charge density of the polyanion, or when using transition metal-substituted POMs with free coordination positions on the metal ions.60-62
The conjugation and the assembly of POMs and peptides is an emerging field of investigation, with promising applications not only in drug design,63 but also in the development of artificial enzymes64 and nanomaterials,65 and in protein crystallography.66 Moreover, the decoration of POM framework with peptides may represent a valuable strategy to investigate specific POM-proteins interactions and to design molecular models of bio-hybrid metal-oxide surfaces or nanoparticles.
With respect to a previously reported research work on the integration of the Anderson-Evans molybdate in a peptide chain,57 we present herein a different Anderson POM-peptide hybrid, obtained combining solid phase synthesis of a small, biologically active peptide with the solution bis-functionalization of the POM. In this article we describe the functionalization of an Anderson molybdate with a Bombesin antagonist peptide (Demobensin-1), as an example of cancer receptor-specific moiety, which was successfully employed to improve the targeting ability of different nanomaterials, such as polymeric NPs,67 metal oxides,68 and gold nanostructures.69, 70 The impact of the interactions between the inorganic and biochemical domains on the structural features and the self-assembly behavior has been evaluated by different analytical techniques (circular dichroism [CD], 2D NMR spectroscopy, dynamic light scattering [DLS] and Z-potential, transmission electron microscopy [TEM]), enabling an advancement in the study of POM-induced peptide folding. The biological activity of the resulting conjugate has been finally discussed, underlying some critical issues related to the use of such kind of bio-hybrids.
2 MATERIALS AND METHODS
Fmoc-protected ethyl indole AM resin was purchased from Novabiochem (Milan, Italy). HATU (O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate), HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate), HOBt (1-hydroxybenzotriazole), DIPEA (N,N-diisopropylethylamine) and TFA (trifluoroacetic acid) were obtained from IRIS Biotech (Marktredwitz, Deutschland). Na2MoO4·2H2O, (CH3CO2)3Mn·2H2O and nBu4NBr, tris(hydroxymethyl)aminomethane (TRIS), dicyclohexylcarbodiimide (DCC), N-Hydroxysuccinimide (NHS), succinic anhydride were purchased from Sigma-Aldrich (Milan, Italy).
2.1 POM synthesis
2.1.1 Synthesis of [(C4H9)4N]4[Mo8O26] (1)
In a 100 mL flask, 5.05 g (2.09 × 10−2 mol) of sodium molybdate dihydrate (Na2MoO4·2H2O) were dissolved in 12 mL of water.71 In a beaker, 3.39 g (1.05 × 10−2 mol) of tetrabutylammonium bromide (nBu4NBr) were dissolved in 10 mL of water. 7.8 mL of HCl 4 M (3.12 × 10−2 mol) were added to the molybdate solution, and the tetrabutylammonium solution was added under vigorous stirring. The white precipitate was filtered on a fritted funnel under vacuum and washed with water, ethanol, and ether (2 × 10 mL each). Yield: 92% (5.18 g, 2.41 mmol).
FTIR (KBr): 3447 (w, br), 2964 (s), 2934 (s), 2873 (s), 1618 (w), 1479 (s), 1377 (m), 1340 (w), 1147 (w), 950 (s), 923 (s), 908 (s), 851 (s), 804 (s), 663 (s), 560 (w), 501 (w), 409 (w).
ESI-MS (-) (CH3CN): 1908.8 ([M – TBA]–), 834.6 ([M – 2TBA]2–).
2.1.2 Synthesis of [(C4H9)4N]3[MnMo6O18((OCH2)3CNH2)2] (2)
In a 250 mL flask, 3.02 g (1.41 mmol) of [(C4H9)4N]4[Mo8O26], 0.56 g (2.08 mmol) of Mn (III) acetate dehydrate, (CH3CO2)3Mn·2H2O, and 0.60 g (4.92 mmol) of TRIS were added.50 Then, 80 mL of acetonitrile were introduced, and the mixture was heated at 80°C for 24 h. A brown precipitate was removed by filtration, while the orange solution was concentrated by heating under vacuum. A white precipitate was also removed by filtration. The remaining solution was crystallized under diethyl ether atmosphere. Orange crystals of the product were collected after 3 days and washed with acetonitrile and diethyl ether (2 × 5 mL each). Yield: 75% (1.97 g, 1.05 mmol).
FTIR (KBr): 3458 (w), 3288 (w), 2965 (s), 2936 (s), 2876 (s), 1619 (w), 1480 (s), 1385 (m), 1341 (w), 1157 (w), 1133 (w), 1031 (s), 938 (s), 915 (s), 900 (s), 797 (w), 738 (w), 662 (s), 564 (m), 518 (w), 455 (m), 413 (m).
1H-NMR (300 MHz, CD3CN): 0.98 (t, 36 H), 1.38 (m, 24 H), 1.62 (m, 24 H), 3.12 (m, 24 H), 61 (s, 12 H).
ESI-MS (-) (CH3CN): 1639.7 ([M-TBA]–), 1398.5 ([M-2TBA + H]–), 1155.2 ([M-3TBA + 2H]–).
Elemental analysis: calcd. for C56H124MnMo6N5O24: C 35.73%; H 6.64%; N 3.72%; found C 35.47%; H 7.02%; N 3.89%.
UV-VIS (CH3CN): λmax= 215 nm, ɛ = 47870 cm−1 M−1.
2.1.3 Synthesis of [(C4H9)4N]3[MnMo6O18{(OCH2)3CNHCO(CH2)2COOH}2] (3)
[(C4H9)4N]3[MnMo6O18((OCH2)3CNH2)2] (2) (268.35 mg 0.143 mmol) and succinic anhydride (286.90 mg, 2.87 mmol) were dissolved in 5.2 mL of DMF in a 10 mL flask.72 The solution was stirred for 24 h at 50°C. Orange crystals were obtained under ether atmosphere, filtered and washed with ether (2 × 5 mL). The crystallization was repeated after dissolving the crystals in CH3CN.
Yield: 90% (266.76 mg, 0.128 mmol).
FTIR (KBr): 3534 (w), 3281 (w), 3224 (w), 3079 (w), 2965 (s), 2927 (s), 2870 (s), 2523 (w), 1733 (m), 1650 (s), 1575 (m), 1480 (m), 1379 (m), 1265 (m), 1183 (m), 1113 (m), 1082 (m), 1025 (m), 938 (s), 917 (s), 901 (s), 663 (s), 557 (w), 507 (w).
1H-NMR (300 MHz, DMSO d6): 0.90 (t, 36 H), 1.30 (m, 24 H), 1.54 (m, 24 H), 2.34 (s, 4 H), 2.66 (s, 4 H), 3.13 (m, 24 H), 7.50 (s, 2 H), 10.04 (s, 2 H), 65.95 (s, 12 H).
ESI-MS (-) (CH3CN): 2083.0 ([M-H]–), 1838.8 ([M-TBA]–), 1718.6 ([2M-3TBA + H]2–), 1149.8 ([2M-3TBA]3–).
Elemental analysis: calcd. for C73H153MnMo6N8O33 C 38.09%, H 6.70%, N 4.87%; found C 37.82%, H 6.99%, N 4.89%.
UV-VIS (CH3CN): λmax = 200 nm. Shoulder at 243 nm. ɛλ200 = 68970 cm−1 M−1
2.1.4 Synthesis of [(C4H9)4N]3[MnMo6O18{(OCH2)3CNHCO(CH2)2CO(C4H4NO3)}2] (4)
Compound 3 (170.75 mg, 0.082 mmol), DCC (101.62 mg, 0.49 mmol), and NHS (38.08 mg, 0.33 mmol) were dissolved in a 10 mL flask in 2 mL of DMF.58 The solution was stirred for ca. 24 h at room temperature. Then, a white precipitate (dicyclohexylurea) was removed by centrifugation and the orange solution was placed under diethyl ether atmosphere for a first crystallization. The product was then recrystallized from CH3CN and washed with diethyl ether (2 × 10 mL). Yield: 78% (145.10 mg, 0.064 mmol).
FTIR (KBr): 3465 (w, br), 3297 (w), 3221 (w), 3080 (w), 2959 (m), 2935 (m), 2874 (m), 1817 (w), 1781 (w), 1735 (s), 1687 (m), 1653 (w), 1559 (w), 1481 (m), 1381 (w), 1261 (w), 1201 (m), 1153 (w), 1115 (w), 1092 (w), 1064 (m), 1027 (m), 941 (s), 920 (s), 901 (s), 812 (w), 668 (s), 562 (w), 458 (w).
1H-NMR (300 MHz, DMSOd6): 1.10 (t, 36 H), 1.47 (m, 24 H), 1.71 (m, 24 H), 2.95 (m, 16 H), 3.31 (m, 24 H), 7.70 (s, 2 H), 63.46 (s, 12 H).
ESI-MS (-) (CH3CN): 2033.8 ([M-TBA]–), 1923.7 ([2M-3TBA + Na]2–), 1795 ([M-2TBA + H]–), 1204 ([2M-4TBA + Na]3–), 896.5 ([M-2TBA]2–).
Elemental analysis: calcd. for C72H138MnMo6N7O34 C 37.99%, H 6.11%, N 4.31%; found C 36.78%, H 6.46%, N 4.14%.
UV-Vis (CH3CN): 200 nm, 216 nm, shoulder at 234 nm. ɛλ200 = 49375 cm−1 M−1.
2.2 Peptide synthesis
Peptides were synthesized by manual solid phase using Fmoc chemistry in 0.06 mmolar scale. Loading on Fmoc deprotected ethyl-indole AM resin with Fmoc-Leu-OH (0.24 mmol) was obtained using equimolar HATU as coupling reagent in the presence of two-fold molar excess of DIPEA. A three-fold molar excess (0.24 mmol) of Fmoc-amino acids have been used for each coupling step, using HBTU/HOBt as coupling reagent. Reaction times were 40 min. Fmoc group was then removed with 20% piperidine. Coupling yields were monitored on aliquots of peptide-resin either by Kaiser test for the amino groups or by evaluation of Fmoc displacement.73 Peptide was simultaneously detached from the resin and removed of all the side-chain protecting groups of the amino acid residues to yield the desired peptide by treatment with TFA-anisole-triisopropylsilane-H2O (95:2.5:2.0:0.5 v/v) (45 min).
The peptide was purified by preparative RP-HPLC using a Shimadzu LC-8 (Shimazdu, Kyoto, Japan) system with a Vydac 218TP1022, 10µ, 250 × 22 mm column. The column was perfused at a flow rate of 12 mL/min employing a binary gradient system (solvent A: 0.05% TFA in water; solvent B: 0.05% TFA in acetonitrile/water, 9:1 by vol.). The fractions containing the desired peptide were collected and lyophilized.
Analytical HPLC analyses were performed on a Shimadzu LC-10 instrument fitted with a Jupiter C18, 10µ, 250 × 4.6 mm column (Phenomenex, Torrance, CA, USA) using the above solvent system (eluent A: 0.05% TFA in H2O; eluent B: 0.05% TFA in 9:1 v/v CH3CN-H2O), flow rate of 1 mL/min, detection at 216 nm (Supporting Information Figure S6). All peptides showed less than 1% of impurities.
2.3 Synthesis of POM-(demobensin-1)2
22.33 mg (9.8 µmol) of POM-NHS (4) and 19.93 mg of peptide (20 µmol) were dissolved in 1.2 mL of DMF, in the presence of 26.7 µL of DIPEA. The solution was left to stir at room temperature overnight. After ca. 15 h the solution was set up for crystallization with diethyl ether to isolate a light orange precipitate. The precipitate was washed with ether (2 × 10 mL), dried and weighed, in 68% yield (26.81 mg, 6.68 µmol).
Analytical HPLC was performed on a Jupiter C18 10 µm 300A 250 × 4.6 mm (Phenomenex, Inc.) column, eluted with (A) 0.05% TFA in H2O, and (B) 0.05% TFA in 9:1 v/v MeCN-H2O; linear gradient from 5% B to 75% B in 70 min; detection at 216 nm (Supporting Information Figure S6).
FTIR (KBr): 3293 (br, m), 3063 (w), 2965 (w), 2929 (w), 2867 (w), 1659 (s), 1534 (m), 1450 (w), 1388 (w), 1346 (w), 1248 (w), 1150 (w), 1115 (w), 1051 (w), 1023 (w), 943 (m), 920 (m), 900 (m), 746 (w), 669 (s).
ESI-MS(-) (CH3CN): 1765.2 ([M – 2TBA]2–), 1655 ([M – 3TBA + Na]2–), 1645.1 ([M – 3TBA + H]2–), 1095.3 ([M – 3TBA]3–).
Elemental Analysis: calcd. for C162H266MnMo6N31O46 C 48.46%, H 6.68%, N 10.82%; found: C 42.81%, H 6.14%, N 10.38%.
UV-Vis (CH3CN): 200 nm, 214 nm and 271 nm (shoulders). ɛλ200 = 143806 cm−1 M−1 (Figure S7).
2.4 Mass spectrometry
Molecular weights of the compounds were determined by ESI-MS on a Mariner (PerSeptive Biosystem, Cambridge, MA, USA) or on a LC/MSD Trap SL, (Agilent Technologies, Santa Clara, CA, USA) mass spectrometers. The mass of the peptide was assigned using a mixture of neurotensin, angiotensin, and bradykinin at a concentration of 1 pmol/μL as external standard.
2.5 DLS and Z-potential
The aggregation of POM-(Demobensin-1)2 was monitored by DLS and Z-potential using a Zetasizer nano series Instrument (Malvern, UK), using low volume disposable plastic cuvettes, upon addition of an increasing amount of stock DMSO solution of POM (6.7 mM), in Milli-Q water. The concentration range was 0.07-0.2 mM. Other samples were only analyzed as 0.2 mM solutions in H2O with 3% DMSO. Z-potential was also monitored in such conditions. In all cases, 3 measurements were collected at 25°C, with automatic selection of number of runs (ranging from 10 to 100) per measurement.
2.6 Circular dichroism

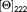 is the detected mean ellipticity per residue at 222 nm and
is the detected mean ellipticity per residue at 222 nm and
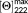 is the maximal theoretical ellipticity at 222 nm calculated from the relationship
is the maximal theoretical ellipticity at 222 nm calculated from the relationship

2.7 1H-NMR
NMR samples were prepared by dissolving appropriate amounts of peptide, and hybrid peptide-POM construct in DMSOd6 (100% isotopic purity, Armar AG, Gottingen, CH) to make approximately 2 mM solutions.
All spectra were run on a Bruker DMX-600 instrument (Bruker Corp., Billerica, MA, USA), operating a 599.90 MHz for 1H. Proton chemical shifts, in parts per million (ppm), are referenced to the residual 1H-DMSO solvent signal (δ = 2.49 ppm). All NMR experiments have been carried out at temperature of 298 K.
One-dimensional (1D) NMR spectra were acquired using typically 16–32 scans with 32K data size. For the two-dimensional (2D) experiments, pulse programs of the standard Bruker library were used. With the exception of COSY experiment, all 2D experiments were acquired in the phase-sensitive mode, with quadrature detection in both dimensions, by use of the time proportional phase increments.75 Typically 512 experiments of 48 scans each were performed: relaxation delay 1 ; size 2K; 6024 Hz spectral width in F2; zero filling to 1K in F1; square cosine or Gaussian multiplication was used in both dimensions before the Fourier transformation. Mixing time of 70 ms was used for TOCSY.76 ROESY77 experiments were run at mixing times of 300 ms. NOESY78 experiments were run at mixing times of 250 ms.
2.8 Transmission electron microscopy
TEM measurements were obtained with a FEI Tecnai G2 transmission electron microscope (Thermo Fisher Scientific, Waltham, MA, USA), operating at an excitation voltage of 100 kV, on 0.2 mM samples dissolved in H2O with 3% DMSO and drop casted on copper grids (400-square mesh). The aqueous solution of the peptide was negatively stained with a drop of 1% (w/v) uranyl acetate solution.
2.9 Cytotoxicity
Cytotoxicity/viability test was performed as 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide (MTT) assay. For the experiments we used HeLa cervical cancer and MCF-7 breast adenocarcinoma cells. HeLa cells show a moderate and MCF-7 breast a high overexpression of the bombesin receptor.79
On day 1, 10 000 HeLa cervical cancer cells/well in a volume of 200 µL were plated in 96-wells plates. For each plate, the first row of wells contained the control cells growth in DMEM medium with 10% FBS (control), and the next contained cells exposed for 2, 24, and 48 h to concentrations of POM or POM functionalized with Demobensin-1 (5, 25, 50, 100, 150 µM) and for the peptide alone (50, 100, 500, 1000, 1500 µM). We tested 2 preparations of the peptide and cells treated with DMSO and TRIS buffer served as control. The results were expressed as the mean ± SD percentage of living cells normalized to the control.
3 RESULTS AND DISCUSSION
The Anderson POM 1 has been initially bis-functionalized with TRIS to obtain [(C4H9)4N]3[MnMo6O18((OCH2)3CNH2)2] (2).50 The amino groups on 2 were then reacted with excess succinic anhydride (20 folds), in DMF at 50°C, for 24 h.72 The product 3, obtained with 90% yield, displays spaced carboxylic groups, which can be available for the reaction with the terminal amino groups of the peptide upon activation with NHS.58 Each step has been monitored with FT-IR, which shows the retention of the spectral features ascribed to the inorganic scaffold, in the region between 500 and 1000 cm−1, in particular MoO (938, 917, 901 cm−1) and MoOMo (663 cm−1) stretching (Supporting Information Figure S1).50, 51 In addition, 3 shows a signal at 1729 cm−1, because of carboxylic CO groups involved in hydrogen bonds with analog molecules, and the absorption of the amidic CO group at 1650 cm−1. 1H-NMR of 3 displays the 2 peaks (2.34 ppm e 2.66 ppm) belonging to the succinic CH2, together with the signal at ca. 66 ppm, because of 12 equivalent CH2 protons close to the paramagnetic Mn3+. ESI-MS(-) shows a main peak at m/z = 1838.8, corresponding to the monovalent POM (m/z for [C48H96MnMo6N4O30]– = 1840). The UV-Vis shows a band at 200 nm (ɛλ200 = 6.89 × 104 cm−1 M−1) LMCT pπ(Ot)→dπ*(Mo), blue shifted with respect to that of the precursor 2 (λmax= 215 nm).49
The activation of 3 is performed with NHS, in the presence of DCC (POM: DCC: NHS = 1:6:4), in DMF, at room temperature for 24 h. The activated POM is then isolated upon crystallization in diethyl ether atmosphere with 78% yield. FTIR highlights the presence of NHS CO vibrational bands at 1817 and 1781 cm−1. In the 1H-NMR, a signal at 2.95 ppm is in agreement with the occurrence of 16 protons in α to the carbonyls, while the other 12 protons resonate at 63.46 ppm. ESI-MS(-) shows the signal of the monovalent compound, at m/z = 2033.8 (m/z calcd. for [C56H102MnMo6N6O34]– = 2034), and of the divalent one at m/z = 896.5 (m/z calcd. for [C40H66MnMo6N5O34]2– = 896). The UV-Vis shows absorptions at 200, 216 nm, and a shoulder at 234 nm (ɛλ200 = 4.94 × 104 cm−1 M−1).
3.1 Peptide design and synthesis
The gastrin releasing peptide receptor (GRP-R) is characterized by the presence of 4 different receptor subtypes (BB1 to BB4) that are overexpressed on a large number of cancers including pancreas, prostate, gastrointestinal, breast, and small cell lung cancers.80 Bombesin, a peptide originally isolated from frog skin and having the same C-terminal amino acid sequence of GRP (Trp-Ala-Val-Gly-His-Leu-Met-NH2), shows marked binding affinity and specificity to the BB2, BB3 and BB4 subtype receptors.81, 82 These findings and others offer impetus to study novel POM conjugates based on bombesin analogs.
A recent comparative study of agonist-peptides and antagonist-peptides indicates that GRP-R antagonists may be superior targeting agents to GRP-R agonist ligands.83 With this aim, structural modifications at the C-terminus of bombesin to yield peptides that retain their binding affinity for GRP receptors conferring antagonist properties, have been performed.84, 85 In particular, the [D-Phe6, Leu-NHEt13, des Met14] BBN[6–14] peptide, named Demobensin-1, lacking of the readily oxidized Met14 residue and containing an N-ethylamide group, is an antagonist designed for targeting GRP receptors,86 which exhibits a much higher in vivo uptake than several agonists. Moreover, the presence of a D-Phe residue in position 6 increases the resistance to proteolysis and consequently the half-life in plasma.
In this study, the H-D-Phe6-Gln-Trp-Ala-Val-Gly-His-Leu-NHEt peptide was synthesized by manual solid-phase on an Fmoc-ethyl indole AM resin that allows to introduce ab initio the C-terminal N-ethylamide group. The POM-(succinyl-D-Phe6-Gln-Trp-Ala-Val-Gly-His-Leu-NHEt)2 construct was synthesized using conventional solution peptide synthesis strategies according to Scheme 1. POM-disuccinyl-N-hydroxysuccinimide esters (4) were dissolved in DMF and added to a deprotected Demobensin-1 solution (2.2 equivs with respect to the POM), in the presence of DIPEA. After 24 h, the solvent was removed under vacuum and the desired product was isolated by crystallization with diethyl ether, with no further purification required, in 68% yield. By FTIR it is possible to observe the disappearance of the NHS CO vibrational bands at 1817 and 1781 cm−1, while amide CO and NH bands can be observed at 1652 and 1531 cm−1, respectively, together with the POM bands (500–1000 cm−1) (Supporting Information Figure S1).

Scheme of synthesis of POM-(Demobensin-1)2
ESI-MS(-) and NMR identification confirmed the grafting by amidation of the peptide onto the hybrid symmetric POM architecture (Supporting Information Figures S2-S5). All the observed peaks in the ESI-MS spectrum, in particular, are consistent with the proposed structure of POM-peptide as TBA salt: the trivalent species, at m/z = 1095.3 (m/z calcd. for [C114H158MnMo6N28O46]3– = 1096) or the divalent species, at m/z = 1765.2 (m/z calcd. for [C130H194MnMo6N29O46]2– = 1765), together with other peaks because of TBA exchange with a sodium ion or proton were observed (Supporting Information Figure S3).
3.2 Conformational studies
The secondary structure of Demobensin-1 alone and in POM-peptide hybrid was investigated by CD and 2D-NMR spectroscopies. CD, in particular, was used to highlight conformational changes of POM-bounded proteins,24, 87 monitor the inhibition of peptide folding/aggregation3 or to assess chirality transfers to a POM scaffold.59, 88
The CD spectra of Demobensin-1 alone and of POM-peptide construct were acquired in trifluoroethanol (TFE) because of the low water solubility of POM hybrid. As seen in Figure 1, the CD spectrum of Demobensin-1 is characterized by a negative band centered at about 200 nm and a positive large signal in the 210–230 nm range containing the contribute of aromatic side-chain transitions, suggesting the presence of an ensemble of different secondary structure.

Far-UV CD spectra of POM-(Demobensin-1)2 (red), Demobensin-1 (blue), and POM 3 (black) in TFE measured with a Jasco J-715 spectropolarimeter. Response 4 s, data pitch 0.2 nm, 1.0 nm bandwidth, 0.1 cm path length (200 µL)
The CD spectrum of POM-peptide hybrid, recorded in the same conditions, is profoundly modified and is characterized by the presence of a strong positive band at about 190 nm and a negative band at 208 nm with a shoulder at about 220 nm, which suggest the presence of a partially ordered α-helix structure.
The determined mean residue ellipticity at 222 nm for the full length symmetric POM-peptide is −12 654 mdeg·cm2·dmol−1, and the associate helical percentage does not exceed 37%, suggesting that only a part of hybrid molecule adopts a helical arrangement. The helical wheel projection of POM-(Demobensin-1)2 (Supporting Information Figure S8) indicated the presence of an amphiphilic helical structure where the hydrophilic face is formed by POM, Suc, Gly, His, and Trp residues.
The analysis of the structure of POM-peptide hybrid was confirmed by 2D-NMR spectroscopy in DMSOd6. Indeed, the low solubility of molecules in TFE did not allow us to use this solvent also in 2D-NMR study. The proton resonances of peptide alone and in the hybrid construct is reported in Table 1. The assignation of the NH resonance of the D-Phe6 residue of Demobensin-1 in the hybrid construct confirmed that an amide bond formation occurred between peptide and POM (for the free amine in Demobensin-1 no signal would have been detected) (Figure 2).

αN cross peaks region of COSY spectra of Demobensin-1 alone (top) or in POM hybrid construct (down) in DMSOd6
| Demobensin-1 | POM-(Demobensin-1)2 | |||||
|---|---|---|---|---|---|---|
| NH | CHα | Others | NH | CHα | Others | |
| TBA+ | - | - | - | - | - | 3.17, 1.56, 1.31, 0.93 |
| Succ | - | - | - | - | - | 2.42, 2.24 |
| D-Phe6 | - | 4.07 | 3.05, 2.90 | 8.39 | 4.39 | 2.87, 7.22 |
| Gln7 | 8.55 | 4.33 | 1.96, 1.80, 1.61 | 8.53 | 3.90 | 1.75, 1.72, 1.46, |
| Trp8 | 8.30 | 4.56 | 3.11, 2.91, 10.8, 7.69, 7.32, 7.18, 7.06, 6.98 | 7.94 | 4.45 | 3.05,10.5, 7.54, 7.33, 7.05, 6.98 |
| Ala9 | 8.25 | 4.41 | 1.20 | 7.77 | 4.33 | 1.26 |
| Val10 | 7.77 | 4.17 | 1.98, 0.86 | 7.54 | 4.12 | 2.02, 0.86 |
| Gly11 | 8.23 | 3.79, 3.72 | - | 8.20 | 3.73, 3.63 | - |
| His12 | 8.11 | 4.59 | 3.05, 2.99, 7.21, 6.78 | 8.02 | 4.50 | 2.98, 2.94, 6.95, 6.70 |
| Leu13 | 8.08 | 4.20 | 1.56, 1.45, 0.91, 0.85 | 7.95 | 4.17 | 1.47, 1.43, 0.86, 0.80 |
| NHEt | 8.06 | 3.08 | 1.01 | 8.04 | 3.08 | 1.01 |
Furthermore, in the 2D-NOESY spectrum, 2 cross peaks between the CH2 of the succinate (2.42 and 2.24 ppm, respectively) and the NH of the D-Phe6 residue can be noticed (Supporting Information Figure S9). The survey spectra clearly show the absence of conformers as confirmed by both COSY and TOCSY dαN cross peaks (Supporting Information Figures S10 and S11), suggesting a high symmetry in the Demobensin-POM-Demobensin hybrid construct.
The analysis of the 2D-NOESY spectrum revealed the presence of a single α, N (i, i + 4) cross peak, diagnostic of the presence of a α-helix conformation, between the CHα of glutamine residue (3.90 ppm) and the NH of the glycine residue (8.20 ppm) in agreement with the CD results (Figure 1 and Supporting Information Figure S9).
As previously observed for analog disc-like Anderson-Evans POMs interacting with proteins,89 the basic oxygen atoms of the POM scaffold seems to be particularly efficient to interact with either the peptide side-chain via electrostatic (with His residues) or the peptide backbone via hydrogen bonds (with the NH amide groups of the main peptide chain).90, 91 Such interactions may be responsible of a conformational evolution resulting into α-helix stabilization. Interestingly, this effect is herein observed with a shorter chain and with a lower amount of polar aminoacids than those previously reported.58 Moreover, although the secondary structure is lost upon addition of water (Supporting Information Figure S12), it is retained in the presence of a competing solvent as DMSO thus highlighting the higher conformational stability of this conjugate.
3.2.1 Aggregation studies
Owing to the limited solubility of the conjugate in an aqueous solution, we have investigated the involvement of intermolecular interactions in the structural rearrangement. As a confirmation of the possible self-assembly, we have analyzed an aqueous mixture (pH 6.8, with 5% DMSO, T = 25°C) of POM by DLS. The formation of aggregates can be observed even with the lowest amount of added POM, with the dimension of the nanoparticles increasing from ca. 20 nm hydrodynamic diameter, Dh, at 70 μM to ca. 44 nm at 0.1 mM. Moreover, at 0.2 mM, the stability of the nanoparticles is higher enough to obtain well defined nanoparticles with Dh ca. 80 nm and polydispersity of 30 nm, Supporting Information Figure S13).
The TEM shows particles with 50–70 nm diameter featuring a core-shell morphology, prone to coalesce into interconnected structures (Figure 3 and Supporting Information Figure S17, E-F). As a matter of fact, the formation of soft vesicle-like structures is common for hybrid POMs bearing hydrophobic chains and exhibiting amphiphilic surfactant behavior,92, 93 whereby counterions with long alkyl chains prevent a close packing of the hybrid clusters.94 Moreover, the negative Z-potential observed for the particles (–32 mV) suggests that the POM exposes its polyanionic surface (Supporting Information Figure S16), while the peptide chains can be both oriented towards the interior of the nanoparticles, as depicted in Figure 3, to form a layered structure.

Proposed structure of the POM-(Demobensin-1)2 hybrid and possible mode of intermolecular interactions in the assembled state, observed by TEM
On the contrary, a POM 2 0.2 mM solution shows the occurrence of nanoparticles with ca 30 nm diameter (Supporting Information Figure S13). The lower stability of these structures is suggested by an almost undefined Z-potential value, as well as by the formation of irregular and elongated structures observed by TEM (Supporting Information Figure S17C). The POM 3 is instead scarcely aggregated in solution, since the particles with Dh 0.67 nm can be ascribed to single POM units (Supporting Information Figure S14). On the other hand, nanoparticles with ca. 10 nm diameter can be observed after deposition on the TEM grid (Supporting Information Figure S17D).
3.3 Biological activity
The aggregation of hybrid POMs bearing lipophilic pendants, in the form of vesicles, was reported to enhance cell internalization, thanks to a higher affinity towards the cell membrane.44 The toxicity and efficacy of POM, POM-(Demobensin-1)2 and the peptide alone were thus tested on HeLa cells (Figure 4 and Supporting Information Figures S18-S20). At the highest tested concentration of 150 µM we found a 73% reduction in cell viability after exposure to both the POM alone and the Demobensin-functionalized POM for 48 h (Figure 4 and Supporting Information Figures S19 and S20). The IC50 was in both cases around 75 µM. In contrast, the peptide alone required a much higher concentration. Here the IC50 was around 1 mM (Supporting Information Figure S18). The result that the IC50 with and without peptide is the same for POM is somehow surprising as HeLa cells are known to overexpress the bombesin receptor to which the Demobensin-1 should target the POM.79 Moreover, even in MCF-7 cells which highly overexpress the receptor (Supporting Information Figure S21) no difference in uptake/cytotoxicity was observed. In this case, an even lower concentration dependent toxicity of POM was detected in the MTT test. This indicates that the molecule is entering the cell in a nonreceptor mediated way. A possible explanation is that the POM influences the correct presentation of the peptide, since the aggregated state of the POM conjugate leads to the embedding of the peptide in the nanoparticles, thus favoring a receptor-independent uptake. On the other hand, the similar activity of the 2 POMs speaks in favor of a similar internalization process, involving a major role of the POM scaffold.44

MTT test on HeLa cells (with a moderate GRP-receptor overexpression) after 48 h incubation with POM 2, Demobensin-1 and the POM-peptide
4 CONCLUSIONS
As mentioned above, there are several explanations for the inability of the peptide to target the receptor. Moreover, considering the size of Demobensin-1 (8 residues) we did not expect it alone to manifest any regular secondary structure, whereas the presence of the molecular metal oxide fosters a specific peptide folding. This conformational effect of POMs, which was previously described by Yvon et al.58 in hybrid Mn-Anderson-peptide construct, confirms that the incorporation of an inorganic POM unit into a short peptide sequence can result in significant new structural features. The partial folding (>30%) into α-helix appears stable and likely involves an interplay of intra and intermolecular interactions between the Demobensin-1 and the POM scaffold.
The new structural features of the conjugate and its self-assembly behavior observed in aqueous solution adversely affect the interaction between the peptide and the GRP receptor. Nevertheless, the POM still retains its biological activity. Within this scenario, it will be interesting to use assemblies of such conjugates as colloidal nano-containers for the delivery of additional drugs or fluorescent tags for imaging studies.65 Moreover, owing to the capability of the POM to direct the rearrangement of a peptide structure,3 intriguing perspectives can be envisaged in the design of semi-synthetic enzymes, in which the POM could act as a catalytic co-factor embedded in a protein-like environment.
CONFLICT OF INTEREST
None of the authors has any financial/conflicting interests to disclose.

