Establishment and characterization of a telomerase-immortalized porcine bronchial epithelial cell line
Abstract
Primary porcine bronchial epithelial cells (PBECs) are an ideal model to study the molecular and pathogenic mechanisms of various porcine respiratory pathogens. However, the short lifespan of primary PBECs greatly limit their application. Here, we isolated and cultured primary PBECs and established immortalized PBECs by transfecting primary PBECs with the pEGFP-hTERT recombinant plasmid containing human telomerase reverse transcriptase (hTERT). Immortalized PBECs (hTERT-PBECs) retained the morphological and functional features of primary PBECs as indicated by cytokeratin 18 expression, telomerase activity assay, proliferation assays, karyotype analysis, and quantitative reverse-transcriptase polymerase chain reaction. Compared to primary PBECs, hTERT-PBECs had higher telomerase activity, extended replicative lifespan, and displayed enhanced proliferative activity. Moreover, this cell line is not transformed in vitro and does not exhibit a malignant phenotype in vivo, suggesting that it can be safely used in further studies. Besides, hTERT-PBECs were susceptible to swine influenza virus of H3N2 subtype and porcine circovirus type 2. In conclusion, the immortalized hTERT-PBECs represent a valuable in vitro model, which can be widely used in the study of porcine respiratory pathogenic infections.
1 INTRODUCTION
Porcine respiratory disease has a great impact on the pig industry worldwide (Hansen et al., 2010) and is still a major health threat for swine producers today (Brockmeier et al., 2017). A recent survey conducted in the United States revealed that respiratory problems are major causes of mortality in swine herds with 53.7% of nursery pig deaths and 60.1% of grower finisher pig deaths attributed to porcine infectious respiratory disease (Nicholson et al., 2012). The porcine respiratory disease may be caused by numerous bacterial and viral agents. The following are considered to be the causative agents of porcine respiratory diseases: Viruses such as porcine circovirus type 2 (PCV2; Huang et al., 2017; P. Zhang et al., 2018) and swine influenza virus (SIV; Krishna et al., 2015; Qiu, De Hert, & Van Reeth, 2015); bacteria such as Bordetella bronchiseptica (Nicholson et al., 2012, 2017) and Streptococcus suis (Zheng et al., 2017); other infectious pathogenic microorganisms such as Mycoplasma hyopneumoniae (Pieters, Daniels, & Rovira, 2017); and lung parasites (García-González et al., 2013). To fully understand the molecular and pathogenic mechanisms of various porcine respiratory pathogens, the development of an appropriate in vitro infection model is required.
The host’s innate immune response is critical to resist respiratory pathogen infection, which plays a pivotal role in safeguarding the airways from inhaled substances (Vos et al., 2005). Airway epithelium, including the bronchial epithelium, has been demonstrated to be involved in the pathogenesis of respiratory diseases in humans and animals (Abraham et al., 2011). Moreover, respiratory epithelial cells initiate and enhance the innate immune response by forming and releasing proinflammatory or anti-inflammatory mediators that can alter cell differentiation, chemotaxis, and immune cell activation (Mayer, Bartz, Fey, Schmidt, & Dalpke, 2008; Xie et al., 2015). Thus, respiratory epithelial cells have traditionally been used as an in vitro model in studies of the pathogenesis of human or murine respiratory pathogen infections (Massin et al., 2010), and they have served as the main target of pathogens in vitro. In the study of porcine respiratory pathogens, including M. hyopneumoniae (Blanchard et al., 1992), SIV (Punyadarsaniya et al., 2011), and S. suis (Meng, Wu, Seitz, Herrler, & Valentin-Weigand, 2016), primary porcine bronchial epithelial cells (PBECs) have been widely used because they provide a close in vitro representation of the airway epithelium. M. hyopneumoniae infection destructs mucosa near the bronchial tissue, resulting in a greater emergence of lymphatic hyperplasia and neutrophil aggregations compared with the trachea (Blanchard et al., 1992). Moreover, previous studies have demonstrated that primary porcine bronchial epithelial cells are more susceptible to the cytotoxic effects of S. suis compared with tracheal epithelial cells (Meng et al., 2016). These studies suggest that it is important to obtain substantial and reliable primary PBECs for the study of porcine respiratory pathogens.
Primary cells isolated from tissues are the major source of epithelial cells, and these primary cells are normal and suitable for future studies of respiratory pathogenicity. However, similar to investigations in primary murine (H. C. Lam, Choi, & Ryter, 2011), human (Vos et al., 2005), canine (Xie et al., 2015), and equine bronchial epithelial cells (Abraham et al., 2011), the primary cultures undergo fairly rapid replicative crisis and can only be propagated for usually no more than six passages (Hahn, 2002; Jiang et al., 2010; Parikh, Nagarajan, Sei-ichi, Sinha, & Garrett-Sinha, 2008). All of these inherent shortcomings of primary cells, including finite proliferation activity, together with the limited amount of isolated tissues present an urgent need for a high proliferation capacity or “immortalized” cell line that retains the ability of an extended lifespan to allow propagation for numerous passages (Miyazawa et al., 2010; Xie et al., 2015). Although there have been several reports on the immortalization of porcine cells, including the establishment of an immortal porcine intestinal epithelial cell line (Wang et al., 2014), immortal neuronal progenitor cells derived from the porcine olfactory bulb (Uebing-Czipura, Dawson, & Scherba, 2008), and immortal porcine luteal cells (L. Zhang et al., 2017), no reliable PBEC lines have been established to date.
In this study, primary PBECs were isolated and cultured. The human telomerase reverse transcriptase (hTERT) gene was then successfully introduced into primary PBECs, resulting in stable hTERT expression. After screening and identification, the immortalized porcine respiratory cell line, designated hTERT-PBECs, was established. These immortalized hTERT-PBECs not only retained the morphological and functional characteristics typical of primary PBECs but extended the lifespan of the cells without causing cancer-associated changes or altering phenotypic properties. Thus, these cells can be used as an appropriate in vitro cell model for studies of different porcine pathogenic infections.
2 MATERIALS AND METHODS
2.1 Experimental animals
One snatch-farrowed, porcine-colostrum-deprived large Yorkshire piglet (7-week-old male) and nine BALB/c nu/nu mice (4-week-old females) were purchased from Zhoubang Biological Technology Company of Nanjing and Animal Experiment Center of Yangzhou University, respectively. The piglet had neither sign of cardiopulmonary disorders nor respiratory symptoms at clinical examination. In addition, the piglet was not under any drug treatment or vaccinated. Before the animal experiment, antibody detection test results based on serologic testing demonstrated that the following pathogens were free, including M. hyopneumoniae, swine fever virus, porcine respiratory and reproductive syndrome virus, pig o-type foot-and-mouth disease, PCV, porcine pseudorabies and SIV.
2.2 Ethics statement
All animal experiments were performed according to animal welfare standards and were approved by the Ethical Committee for Animal Experiments of Jiangsu Academy of Agricultural Sciences, China.
2.3 Isolation and culture of primary PBECs
The primary PBECs were isolated according to the method for mouse tracheobronchial epithelial cells (H. C. Lam et al., 2011) and human epithelial cells from lungs (Ramirez et al., 2004; Vos et al., 2005) with some modifications. The piglet was euthanized with an overdose of 20% sodium pentobarbital (2 ml/kg; Cat. #P3761; Sigma-Aldrich, St. Louis, MO) through forelimb intravenous injection to ensure it suffered minimal pain. Directly after euthanasia, 75% alcohol was used to disinfect the epidermal tissue of the thoracic and abdominal skin of the piglet. The skin was cut along the sternum from the throat to the last rib until the rib cage was removed to expose the chest. Another set of surgical scissors and scalpel were used to remove most of the skin around the tracheal in case of throat contamination. Subsequently, the trachea was lifted after connective tissue was removed. The trachea distal to the lung was cut immediately after the upper trachea was fastened with a hemostatic clamp. The remaining trachea together with the intact lung tissue were removed from the chest and placed in a sterilized tray covered with ice-cold phosphate-buffered saline (PBS) before being transferred to the biosafety cabinet. For further bronchial separation, the lung tissue was divided into single lobes with flattened tweezers. Subsequently, a surgical dissecting mirror, microsurgical forceps and scissors were used to remove the excess lung tissue surrounding the trachea, eventually leading to the isolation of bronchial tissue.
The isolated bronchial tissues were washed with ice-cold D-Hank’s balanced salt solution (DBSS; Ca2+/Mg2+ free; Gibco, Carlsbad, CA) and manually cut into approximately 1 mm3 pieces with microscopic scissors. The minced bronchial tissue pieces were digested in a total of 20 ml of digestion solution for 30 min at 37°C in a 5% CO2 atmosphere. The digestion solution consisted of Dulbecco’s modified Eagle's media–nutrient mixture F-12 (DMEM–F12), containing 4 ml of ethylenediaminetetraacetic acid (0.0005 M), 8 ml of collagenase I (0.5 mg/ml; Worthington, Lakewood, NJ), and 8 ml of 0.25% trypsin (Gibco, Carlsbad, CA). Twenty microliters of the solution was removed to observe the digestion condition under microscopy every 20 min. Ice-cold DMEM–F12 medium containing 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA) was added to stop the digestion immediately when the cells become round and scattered. The crude cell-containing suspension was filtered through a 200-mesh filter after gentle pipetting. The filtrate was collected after the filter was rinsed twice with ice-cold DBSS. The supernatant was discarded after centrifugation of the suspension at 1200 rpm for 10 min, and the precipitation was resuspended with DMEM–F12 containing 10% FBS before being placed into four 10 cm dishes (Thermo Fisher Scientific, Waltham, MA) and two wells of 12-well cell plate (Corning, Tewksbury, MA). Cultures were grown in DMEM–F12 medium containing 100 U/ml penicillin (HyClone, Marlborough, MA), 0.1 mg/ml streptomycin (HyClone, Marlborough, MA), 15 μg/ml enrofloxacin (Aladdin, Shanghai, China), 0.1 mg/ml gentamicin, and 5 μg/ml amphotericin (Dingguo, Beijing, China). To get the primary PBECs, the medium in four dishes was changed to DMEM–F12 containing 2% FBS after 12 hr to restrict the outgrowth of nonepithelial cells, such as smooth muscle cells. In addition, PBEC cultures were supplemented with antibiotics and regulation factors of epithelial cells (Cat. #CC-4175; LONZA, Walkersville, MD), including 26 µg/ml bovine pituitary extract, 15.5 ng/ml human recombinant epidermal growth factor (hEGF), 5 µg/ml human recombinant insulin, 1.4 µM hydrocortisone, 2.7 µM epinephrine, 9.7 nM tri-iodo-l-thyronine, 0.3 nM retinoic acid, and 10 µg/ml transferrin. Culture medium was changed every day, and an additional 10 ng/ml hEGF and 0.1 ng/ml retinoic acid (Sigma-Aldrich, St. Louis, MO) were added each day. To get the primary smooth muscle cells used as control, the supernatant of cells in 12-well cell plate was changed to DMEM–F12 medium containing 10% FBS and without adding epithelial cells growth factors after smooth muscle cells demonstrated adherent growth. Then, the growth of primary PBECs could be restricted and primary smooth muscle cells will gradually become the predominant cells. Morphological features of the primary cultures were evaluated every day by light microscopy (Nikon, Chiyoda-ku, Tokyo, Japan). The cells were passaged upon reaching 80% confluence after approximately 3 days in culture by digestion with 0.125% trypsin (Gibco, Carlsbad, CA) diluted in D-Hank’s medium.
2.4 Characterization of primary cell cultures
To determine whether the isolated cells were bronchial epithelial cells, cytokeratin 18 immunostaining was performed according to previous studies (Schierack et al., 2006; Wang et al., 2014; Xie et al., 2015). Specifically, primary PBECs at passage 2 and primary smooth muscle cells containing few primary PBECs at passage 2 (negative control) were grown in a 24-well plate before fixation with 4% paraformaldehyde in PBS for 10 min at room temperature (RT). Subsequently, cells were treated with 0.1% Triton X-100 in PBS for 5 min at RT. Cells were blocked with an immunostaining blocking buffer containing 1% bovine serum albumin (Beyotime, Shanghai, China) for 30 min and then incubated with a 1:600 dilution of primary monoclonal antibody against cytokeratin 18 (Cat. #ab668; RRID: AB_305647; Abcam, Cambridge, MA) for 1.5 hr at 37°C. After three PBS washes, cells were incubated with fluorescein isothiocyanate (FITC)–conjugated goat anti-mouse immunoglobulin G (IgG; H + L; Cat. #A0568; Beyotime, Shanghai, China) at a 1:1,000 dilution for 1 hr at 37°C. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 15 min before cells were observed using fluorescence microscopy (Zeiss, Tokyo, Japan). To assess the percentage of PBECs in isolated primary cells, six microscopic fields at ×100 magnification were randomly selected for each of three wells in a 24-well plate to manually count the cell number using the same cutoff values and the same exposure settings.
2.5 Establishment of immortalized hTERT-PBECs
After culturing for two passages, primary PBECs were plated into 24-well plates at a density of 2 × 105 cells per well and cultured in DMEM–F12 medium containing 2% FBS plus the regulation factors and antibiotics described as above. Before transfection, the medium was changed to fresh medium with FBS and antibiotics. The primary cells were transfected with the pEGFP-hTERT recombinant vector constructed in our previous study (Xie et al., 2015) when the cells reached 80–85% confluence. In addition, the pIRES2-EGFP vector was also transfected as a control using Lipofectamine® 3000 (Invitrogen, Waltham, MA) according to the manufacturer’s instructions. The cells were observed using fluorescence microscopy (Zeiss, Tokyo, Japan) after being in stationary culture for more than 24 hr. pEGFP-hTERT contains green fluorescent protein (EGFP), which was used as a marker to verify the transfection success rate of the recombinant plasmid into primary PBECs. Because pEGFP-hTERT contains the neo gene, which confers resistance to the G418 antibiotic, positive cell clones were selected with 400 µg/ml G418 (diluted in 1 mol/L HEPES; Sigma-Aldrich, St. Louis, MO) in complete culture medium after 24 hr, and monoclonal cells appeared after 6 days of selection. Stable hTERT overexpression cell lines were established from large and healthy G418-resistant colonies. After 24 hr of incubation, the medium was replaced, and the positive cells were selected by treatment with 400 mg/ml G418 antibiotics, and cells were selected a second time using the same procedure. Eventually, positive cell clones were obtained after three rounds of selection. After confirmed as mycoplasma negative, the newly established cell line, hTERT-PBECs, at passage 30 was sent to China Center for Type Culture Collection (CCTCC) for preservation (CCTCC no: 201749).
2.6 Cell growth kinetics and replicative lifespan determination
Unless otherwise stated, primary PBECs and immortalized hTERT-PBECs used in all subsequent experiments are primary PBECs at passage 4 and hTERT-PBECs at passage 60, respectively. The hTERT-PBECs and primary PBECs were seeded in 24-well plates at 2 × 104 cells per well in 500 µl of culture medium. The medium was changed every two days. On each day, cells in three wells were harvested by trypsinization, and cell numbers were counted according to a previous study (Hong, Zhang, Xu, Su, & Sun, 2007; Moffatt-Jauregui et al., 2013). Each test was performed in triplicate.
To determine the replicative lifespan, primary PBECs and hTERT-PBECs were trypsinized and collected after centrifugation. Cells were fixed overnight with 10 ml of 75% ice-cold ethanol at 4°C and then washed three times using cold PBS. After centrifugation, the supernatant was discarded, and cells were washed twice with cold PBS and treated with 0.4 ml propidium iodide (Cat. #550825; BD Biosciences, San Jose, CA) for 15 min in the dark at RT. For proper cell cycle analysis, the count of cells should be more than 1 × 106. The percentages of gated cells in the G1/G0, G2/M, and S phases were calculated. Cell cycle quantification was examined by flow cytometry (FCM) on a BD Accuri C6 (BD Biosciences, San Jose, CA), and data were processed to determine the cell cycle distribution using FlowJo software (Su et al., 2013; Wang et al., 2014).
2.7 Reverse-transcriptase polymerase chain reaction (RT-PCR) analysis of hTERT
Total RNA was extracted from primary PBECs, hTERT-PBECs, and human epithelial type 2 (HEp-2) cells (positive control; CLS Cat. #300397/p694_Hep-2; RRID: CVCL_1906) using a Total RNA Extraction Kit (Cat. #R6834; Omega, Norcross, GA) according to the manufacturer’s instructions. RNA was extracted from approximately 1 × 106 cells mentioned, and complementary DNA (cDNA) was synthesized using an oligo(dT) primer and HiScript® Reverse Transcriptase (Cat. #R101; Vazyme, Nanjing, China). The transcriptional level of hTERT was analyzed by RT-PCR, and β-actin was used as an internal control. The primer sequences of hTERT (forward: 5′-CCGTGGTTTCTGTGTGGT-3′; reverse: 5′-GAAGCGGCGTTCGTTGT-3′) and β-actin (forward: 5′-CACGCCATCCTGCGTCTGGA-3′; reverse: 5′-AGCACCGTGTTGGCGTAGAG-3′) were designed referring to the human and porcine messenger RNA (mRNA) homologous sequence. The amplified PCR products of hTERT and β-actin were 661 and 100 base pairs, respectively.
2.8 Western blot analysis
As previously described (Su et al., 2013), no less than 5 × 106 primary PBECs, hTERT-PBECs, and HEp-2 cells (positive control) were washed twice with PBS and collected before using a total protein extraction kit (Cat. #E211; Vazyme, Nanjing, China). Cell proteins were rapidly frozen in liquid nitrogen and stored at −70°C until protein concentrations were measured. Approximately 15 µg of total protein was resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to a nitrocellulose membrane (Millipore). The membranes were washed three times with PBS and then blocked with 5% nonfat milk (Yili Industrial Group Co., Ltd., Hohhot, China) in Tris-buffered saline (TBS) containing 0.5% Tween 20 (TBST) at 37°C for 2 hr with shaking. Subsequently, the membranes were incubated with anti-hTERT (Cat. #sc-377511; RRID: AB_11150127; Santa Cruz Biotechnology, Dallas, TX) or anti-β-actin (Cat. #BS6007MH; Bioworld, St. Louis, MN) at 1:600 in blocking solution at 37°C for 1.5 hr. After three washes using TBST, the membranes were incubated with secondary antibody (horseradish peroxidase–labeled goat anti-mouse IgG (H + L); Dingguo, Beijing, China) at 1:10,000 in blocking solution at 37°C for 1.5 hr followed by incubation with chromogenic reagents (Tiangen, Beijing, China).
2.9 Telomerase activity assay
Telomerase activity analysis was performed according to a previous study with some modifications (He et al., 2009). Briefly, primary PBECs, hTERT-PBECs, and HEp-2 cells (positive control) were assayed with a TeloTAGGG Telomerase PCR ELISAPLUS Detection Kit (Cat. #11854666001; Roche, Pleasanton, CA) according to manufacturer’s instructions. In addition, HEp-2 cells were treated for 10 min at 85°C (named as H-HEp-2) to inactivate telomerase protein for use as a negative control. The concentrations of cells were adjusted to the same concentration, and cell extracts were separately prepared. The absorbance was determined at 450 nm with a reference wavelength of 690 nm. Each test was performed in triplicate. It was considered there was no telomerase activity when the absorbance was less than 0.2 according to a previous study (He et al., 2009).
2.10 Karyotype analysis
The number of chromosomes in hTERT-PBECs was determined following standard methods (Ha Thi et al., 2014; Wang et al., 2014). Briefly, cells were initially cultured in a T25 cell culture flask (Thermo Fisher Scientific, Waltham, MA) for more than 24 hr, and cells were then treated with colcemid (Aladdin, Shanghai, China) at a final concentration of 0.2 μg/ml at 37°C for 5 hr. Subsequently, cells were trypsinized, centrifuged, and incubated in 0.075 M KCI at 37°C for 20 min. Cells were then fixed with a freshly prepared ice-cold acetic acid–methanol (1:3, vol/vol) solution and subjected to blowing and careful mixing with straws before collection by centrifugation at 1,000 rpm. This step was repeated, and two or three drops of dispersed cell suspension were smeared on each ice-cold slide from a 0.5 m height and then air-dried. The chromosomes were stained with 4% Giemsa solution in Gurr’s buffer, and the number of chromosomes in metaphase (n = 100 cells) was observed microscopically.
2.11 Relative quantitative RT-PCR analysis
Total cellular RNA was isolated from both primary PBECs and hTERT-PBECs after direct cell lysis using the Total RNA Extraction Kit (Cat. #R6834; Omega, Norcross, GA). No less than 1 μg of total cellular RNA was reverse-transcribed in a 10-µl reaction volume using the HiScript II Q RT SuperMix for quantitative PCR (+gDNA wiper; Cat. #R223; Vazyme, Nanjing, China) before running on an ABI 7500 Real Time PCR System using the HiScript II One Step qRT-PCR SYBR® Green Kit (Cat. #Q221; Vazyme, Nanjing, China). RT-PCR was performed using cDNA under specific conditions for a total of 16 genes. These selected genes are involved in cell cycle (sox2, sox17, ccna1, ccna2, ccnb1, and ccnb2; H. L. Chen, Chew, Packer, & Gallo, 2013; Ha Thi et al., 2014; Klajic et al., 2014; Lange, Keiser, Wells, Zorn, & Whitsett, 2009; Tompkins et al., 2011), innate immunity (IL-6, IL-8, CSF2, JNK1, CXCL2, and STAT3; Lamontagne, Mell, & Bouchard, 2016; Machugh et al., 2012; Nalpas et al., 2013), and oxidative stress response (DUOX1, DUOX2, SOD1, and GPX1; Brockmeier et al., 2017; X. Chen et al., 2017; Nicholson et al., 2012). Glyceraldehyde 3-phosphate dehydrogenase and β-actin were used as the internal controls. PCR primers used in the quantitative assays are listed in Supporting Information Table S1. The fold change of mRNA expression in primary PBECs and hTERT-PBECs was determined using the 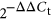 method.
method.
2.12 In vitro soft agar assay
The soft agar assay was performed as previously described (He et al., 2009). Briefly, a bottom layer of 0.5% agar gel with 1.5 ml of culture media was prepared in a 24-well plate and stored overnight at 4°C to solidify the gel. Subsequently, hTERT-PBECs were trypsinized and re-suspended at concentrations of 5 × 103, 1 × 104, and 2 × 104 cells/ml in DMEM–F12 medium containing 2% FBS. One milliliter of cell suspension (final concentration: 0.33% agar) was added into 1.5 ml of 0.5% agar. Cells were overlaid onto the solidified lower layer and incubated at 37°C in a humidified atmosphere with 5% CO2 for 4 weeks. HEp-2 cells served as positive controls, and colonies in the plate were observed under a microscope for over 1 month. The presence of even a single colony indicated that cells were capable of anchorage-independent growth.
2.13 In vivo nude mice tumorigenicity assay
To evaluate the functional tumorigenicity of immortalized cells in vivo, a tumorigenicity assay was performed by subcutaneously injecting hTERT-PBECs into the left flank of 4-week-old nude mice (one site per mouse at 1.5 × 106 cells per mouse for a total of three mice). As a positive control, HEp-2 cells at the same concentration were injected into the flanks of another three nude mice. Animals were housed under specific pathogen-free conditions and were observed weekly for tumor appearance for 2 months before they were killed.
2.14 Indirect immunofluorescence assay
To test the susceptibility of immortalized cells to swine respiratory-related viruses, SIV of H3N2 subtype and PCV2 were selected. Mardin Darby Canine Kidney (MDCK) cells and hTERT-PBECs were both infected and mock-infected with influenza A/swine/Shandong/3/2005(H3N2) at the multiplicity of infection (MOI) of 0.1, and Porcine Kidney 15 cells and hTERT-PBECs were infected and mock-infected with PCV2 isolate Haian (Genbank accession number FJ712216.1) at the MOI of 0.1. The procedure of indirect immunofluorescence assay was performed as previously described (Ding et al., 2017). Briefly, cells mentioned above were plated into 24-well plates at a density of 5 × 104 cells per well in prior night before virus infection. After infected with influenza and PCV for 24 and 48 hr, respectively, cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min at RT. Cells were permeabilized with 0.2% Triton-X100 in PBS for 2 min at RT. After blocking with 5% bovine serum albumin for 2 hr at 37℃, the cells were exposed to positive pig serums for whole virus against influenza and PCV2 produced in our lab at a 1:500 dilution for 1 hr at 37°C. After three times washing with PBS, cells were incubated with FITC-conjugated goat anti-pig IgG (H + L; Cat. #ab6911; Abcam, Cambridge, MA) at a 1:1,000 dilution for 1 hr at 37°C. Cell nuclei were stained with DAPI for 5 min at RT before cells were visualized using fluorescence microscopy (Zeiss, Tokyo, Japan).
2.15 Statistical analysis
Data from quantitative real-time PCR analysis by using ABI 7500 Real Time PCR System (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA) and cell growth curves were analyzed by SPSS Statics software v20.0 developed by (IBM, LLC, Armonk, NY; http://www-01.ibm.com/software/uk/analytics/spss/; RRID: SCR_002865), and cell cycle analysis was analyzed by FlowJo software v7.6 developed by (FlowJo, LLC, Ashland, OR; https://www.flowjo.com/solutions/flowjo; RRID: SCR_008520). Statistical differences were assessed via analysis of variance, and p < 0.01 was considered to be significant.
3 RESULTS
3.1 Isolation, culture, and characteristics of primary PBECs
After the piglet was euthanized and disinfected with 75% alcohol, blood was drained from the abdominal incision, and a throat incision was made to expose the trachea to avoid contaminating the lung tissue. As shown in Figure 1a, all of the larynx and trachea tissues connecting the intact lung tissue were obtained. Isolated tissue was put in a tray before transferring to biosafety cabinet for further bronchus isolation. Figure 1b shows the isolated bronchus in a sterilized glass dish after gentle removal of the entire lung tissue around the trachea with microsurgical forceps and scissors. The isolated bronchial tissues were digested and cultured in medium with 10% FBS, and the supernatant was changed to medium with 2% FBS together with supplemental regulatory epithelial factors. Finally, few colonies of primary PBECs at passage 1 were grown to cell confluency greater than 80%. As shown in Figure 1c, primary cells displayed the cuboidal phenotype typical of mammary epithelial cells and grew in close contact to each other. Cytokeratin 18 immunostaining was performed after passage 1 primary cells were passaged to confirm that the primary cells isolated were indeed bronchial epithelial cells rather than nonepithelial cells, such as smooth muscle cells. The primary PBECs were stained green, and all cell nuclei were stained in blue using DAPI (Figure 1d), and the purity of primary PBECs was 92.6 ± 5.6%. For the negative control, only very limited cells (primary PBECs) were stained in green, whereas the majority cells (primary smooth muscle cell) were only blue-stained within cell nuclei.

Isolation and culture of primary PBECs. (a) The whole larynx and trachea tissue connecting intact lung tissue isolated from piglet before transferred to a biosafety cabinet. (b) Isolated bronchials are shown in a sterilized glass dish after gently removal of the entire lung tissue around the trachea with microsurgical forceps and scissors. (c) Primary culture of PBECs at passage 1 showing few colonies (×100 magnification) and displayed the cuboidal phenotype typical of mammary epithelial cells. (d) Immunofluorescence identification of cytokeratin 18 on primary PBECs (×200 magnification). (e) Immunofluorescence identification of cytokeratin 18 on primary smooth muscle cells with few primary PBECs (×200 magnification). PBEC: porcine bronchial epithelial cell [Color figure can be viewed at wileyonlinelibrary.com]
3.2 Establishment of stable hTERT-PBECs
The primary PBECs could not be cultured for longer than six passages due to their reduced and short proliferative potential. Therefore, primary PBECs were transfected with the pEGFP-hTERT recombinant vector to overexpress hTERT and ultimately establish the stable hTERT-overexpressing cell line, hTERT-PBEC, which could be maintained for more than 60 passages. Because the recombinant plasmid encodes EGFP, it was used as a marker to verify successful transfection of recombinant plasmid by observation with fluorescence microscopy. Fluorescence intensity and quantity of hTERT-PBECs are shown in Figure 2a. Moreover, hTERT-PBECs were also immunostained for the cytokeratin 18 epithelial marker as described above. The hTERT-PBECs retained the characteristics of epithelial cells (Figure 2b), and the purity of hTERT-PBECs at passage 60 was 95.9 ± 3.2%.

Identification of immortalized hTERT-PBECs. The hTERT-PBECs were transfected with the recombinant plasmid pEGFP-hTERT via Lipofectamine 3000. (a) Fluorescence intensity and quantity using fluorescence microscopy at passage 60. (b) Immunostaining of hTERT-PBECs at passage 60 for cytokeratin 18. hTERT: human telomerase reverse transcriptase; PBEC: porcine bronchial epithelial cell [Color figure can be viewed at wileyonlinelibrary.com]
3.3 Extended replicative lifespan of hTERT-PBECs
The proliferation potential of immortalized hTERT-PBECs was assessed based on their growth, morphological characteristics, and cell cycle analysis. Growth curves of both primary PBECs and hTERT-PBECs are shown in Figure 3a. Both cell types showed a rapid increase in cell proliferation 48 hr after passage. However, the concentration of primary PBECs was higher than the immortalized hTERT-PBECs from Days 3 to 5 (without significant difference), and the primary cells reached a maximal growth rate at Day 5 before declining. In contrast, hTERT-PBECs reached their peak at Day 7, and the concentration of the hTERT-PBECs was higher than that of primary PBECs from Days 7 to 9 (p < 0.01). However, no obvious differences were found between the morphological characteristics of the primary PBECs (Figure 3b) and the immortalized hTERT-PBECs (Figure 3c). To ascertain whether hTERT expression affected cell replicative lifespan or cell cycle progression, cell cycle distribution was assessed by FCM. Several differences were observed in the cell cycle phases between primary PBECs (Figure 3d) and hTERT-PBECs (Figure 3e) under the same culture conditions. The percentage of hTERT-PBECs in phase S was 11.55% higher than that of primary PBECs, whereas the percentage of hTERT-PBECs in phase G1 was lower than that of primary PBECs. All the above results indicated that the hTERT-PBECs have an extended replicative lifespan and display enhanced proliferative activity compared to primary PBECs.

Comparison of the cell proliferation capacity of primary PBECs and immortal cells after hTERT transfection. (a) Growth of hTERT-PBECs at passage 60 and control primary PBECs at passage 4. The data are representative of three independent experiments. **p < 0.01 indicates significant differences between the two groups at Days 7, 8, and 9. (b) Morphological characteristics of primary PBECs at passage 4. (c) Morphology of the immortalized cells at passage 60. (d) Cell cycle distributions of primary PBECs at passage 4. (e) Cell cycle distributions of immortalized hTERT-PBECs at passage 60. The percentages of primary and immortalized cells in the S phase were 3.46% and 15.01%, respectively. hTERT: human telomerase reverse transcriptase; PBEC: porcine bronchial epithelial cell [Color figure can be viewed at wileyonlinelibrary.com]
3.4 Increased telomerase activity of hTERT-PBECs
As demonstrated in Figure 4a, hTERT was expressed in hTERT-PBECs and HEp-2 cells (positive control), but hTERT was not expressed in primary PBECs. Whole-cell extracts of primary PBECs, hTERT-PBECs, and HEp-2 cells were collected and subjected to western blot analysis. hTERT was detected in hTERT-PBECs and HEp-2 cells but was not observed in primary PBECs (Figure 4b). To determine the telomerase activity of primary PBECs and hTERT-PBECs, telomeric replication amplification protocol (TRAP) was performed. As depicted in Figure 4c, hTERT-PBECs showed significantly higher telomerase activity than that of primary PBECs. The absorbance of immortalized hTERT-PBECs was 0.85, whereas the absorbance primary PBECs was 0.15 (<0.2). The absorbance of the H-HEp-2 heat-inactivated negative control sample was 0.03, while the absorbance of the HEp-2 positive control was 0.89. These results suggested that primary PBECs do not express telomerase activity and that the telomerase activity of hTERT-PBECs was relatively higher.

Characteristics of immortalized hTERT-PBECs. (a) hTERT messenger RNA expression tested by reverse-transcriptase polymerase chain reaction (661 bp). Lane M, DNA marker DL5000; lane 1, primary PBECs at passage 4 as a negative control. lane 2, hTERT-PBECs at passages 60; lane 3, a laryngeal cancer cell line (HEp-2), as a positive control. β-actin, 100 bp, was used as an internal control. (b) hTERT western blot analysis. The molecular weight of hTERT was 123 kDa. Lane M, protein prestained mass markers; lane 1, primary PBECs at passage 4 as a negative control; lane 2, hTERT-PBECs at passage 60; lane 3, HEp-2 cells used as a positive control. β-Actin, 42 kDa, was used as an internal control. (c) Telomerase activity of immortalized hTERT-PBECs and control PBECs evaluated by TeloTAGGG Telomerase PCR ELISAPLUS Detection Kit. HEp-2 and heat-inactivated HEp-2 samples were used as positive and negative controls, respectively. HTERT-PBECs had higher telomerase activity than primary PBECs (p < 0.01). Datawere given as mean of three experiments with n = 3 for determination telomerase activity. **p < 0.01. bp: base pair; HEp-2: human epithelial type 2; hTERT: human telomerase reverse transcriptase; PBEC: porcine bronchial epithelial cell [Color figure can be viewed at wileyonlinelibrary.com]
3.5 Stable chromosomes of immortalized hTERT-PBECs
To analyze the karyotype stability of the immortalized hTERT-PBECs, actively proliferating hTERT-PBEC cultures at passage 60 were selected. As depicted in Figure 5a, hTERT-PBECs showed a typical and representative karyotype as well as a normal porcine diploid chromosome number (2n = 38). The chromosomal rearrangements are shown in Figure 5b, including 18 pairs of autosomes and one pair of sex chromosomes, which was consistent with the male sex of the piglet.

Karyotype analysis of immortalized hTERT-PBECs. (a) Karyotype analysis of hTERT-PBECs at passage 60. (b) Chromosomal rearrangements of hTERT-PBECs at passage 60, indicating that the cells contained 38 chromosomes and isolated from male piglet, consistent with a diploid karyotype. hTERT: human telomerase reverse transcriptase; PBEC: porcine bronchial epithelial cell [Color figure can be viewed at wileyonlinelibrary.com]
3.6 Gene expression analysis in primary PBECs and hTERT-PBECs
To investigate if hTERT introduction affected gene expression, relative quantitative RT-PCR was performed in both primary PBECs and immortalized hTERT-PBECs. Figure 6 shows the expression levels of 16 functional genes. The expression levels of the genes involved in the cell cycle, namely, sox2, ccna1, ccna2, ccnb1, and ccnb2, were significantly upregulated in immortalized hTERT-PBECs compared with primary cells, except for sox17, which was upregulated by only a 0.5-fold change. In contrast, the expression levels of genes involved in immune response and oxidative stress response remained unchanged as the relative mRNA levels ranged from 0.56 to 1.64 indicating less than a two-fold change, which is often used as a criterion for distinguishing significantly differentially expressed genes (Lamontagne et al., 2016; Nalpas et al., 2013).

Relative expression levels of genes involved in cell cycle, innate immunity, and response to oxidative stress in immortalized hTERT-CBECs compared to the primary cells (set to 1). Gene expression was determined by quantitative real-time PCR analysis. Error bars represent standard deviations from three independent experiments. hTERT: human telomerase reverse transcriptase; PBEC: porcine bronchial epithelial cell
3.7 Immortalized hTERT-PBECs are not transformed in vitro and do not exhibit a malignant phenotype in vivo
To confirm if immortalized hTERT-PBECs could lead to cellular anchorage-independent growth in vitro, a soft agar colony formation assay was performed. Several colonies were observed in the wells of positive control HEp-2 cells even at the lowest cell density of 5 × 103 cells per well (Figure 7a), but no colonies were observed in the wells of hTERT-PBECs even after 4 weeks (Figure 7b). To determine whether immortalized hTERT-PBECs are tumourigenic in vivo, a definitive functional assay for tumorigenicity was performed by subcutaneously injecting hTERT-PBECs, primary PBECs, or tumourigenic HEp-2 cells into nude mice. Two months later, tumors were observed in all the three mice injected with HEp-2 cells (positive control; Figure 7c). In contrast, none of the mice injected with hTERT-PBECs (Figure 7d) or primary PBECs (negative control; data not shown) developed tumors. Samples were collected from the injection sites for histopathological examination, which confirmed the above observation results. Normal muscle tissue structures were observed at the site of injection of primary cells (data not shown) or immortalized hTERT-PBECs (Figure 7f,h), whereas pyogenic lesions were formed surrounding the tumor tissue. A large number of infiltrating dense inflammatory cells was observed at the HEp-2 injection site (Figure 7e,g). All these results indicated that immortalized hTERT-PBECs are not transformed and do not have tumourigenic potential.

In vitro soft agar assay and in vivo nude mice tumorigenicity assay of immortalized hTERT-PBECs. (a) Soft agar assay of positive control HEp-2 cells at the cell density of 5 × 103 cells/ml, ×100 magnification. (b) Soft agar assay of hTERT-PBECs at passage 60, ×100 magnification. Primary PBECs at passage 4, hTERT-PBECs at passage 60, or positive-control HEp-2 cells were subcutaneously injected into nude mice. (c) Three nude mice inoculated with the positive-control HEp-2 cells, developing tumors at the injection site in 2 months. (e) Histological examination demonstrated a dense inflammatory cellular mass below the HEp-2-cell injection site, ×100 magnification. (g) A dense inflammatory cellular mass was observed below the HEp-2-cell injection site, ×400 magnification. (d) No tumors were observed in three nude mice inoculated with hTERT-PBECs at passage 60. (f) Morphologically normal tissue was observed below the injection site of the hTERT-PBECs at passage 60, ×100 magnification. (h) Morphologically normal tissue was observed below the injection site of the hTERT-PBECs at passage 60, ×400 magnification. HEp-2: human epithelial type 2; hTERT: human telomerase reverse transcriptase; PBEC: porcine bronchial epithelial cell [Color figure can be viewed at wileyonlinelibrary.com]
3.8 Susceptibility of hTERT-PBECs to swine respiratory-related viruses
To test the susceptibility of hTERT-PBECs to SIV of H3N2 subtype and PCV2, indirect immunofluorescence assay was performed. As depicted in Figure 8, microscopic examination showed that immortalized hTERT-PBECs infected with SIV exhibited cytopathic effect (CPE) in some degree such as scattered growth and cell rounding. Similarly, in MDCK-SIV-infected group (positive control), CPE including cell rounding, detachment, cell apoptosis, and death was observed. Cells in SIV mock-infected groups showed normal cell morphology with no obvious CPE. Strong green fluorescent signals were observed in both MDCK cells and hTERT-PBECs infected with SIV, while cells in SIV mock-infected groups demonstrated no specific immunofluorescent signals. For cells infected with PCV2, no CPE was observed in PK15 cells (positive control), while hTERT-PBECs showed kind of CPE including cell rounding and death. However, typical circular green fluorescence signals could be observed in both PCV2-infected PK15 cells and hTERT-PBECs, while no specific green fluorescence signal was observed in PCV2 mock-infected groups. These results suggested that hTERT-PBECs were susceptible to swine respiratory-related viruses such as SIV and PCV2.

Immunofluorescence assay of immortalized hTERT-PBECs infected with SIV and PCV2. Immortalized cells, MDCK cells (positive control for SIV infection), PK15 cells (positive control for PCV2 infection) infected with SIV and PCV2 at the MOI of 0.1, respectively. Cells were stained with positive pig serums of whole virus for SIV and PCV2 at a 1:500 dilution as primary polyclonal antibodies, and FITC-conjugated goat anti-pig IgG (H + L) as secondary antibodies at a dilution of 1:1,000. FITC, antibody reactivity to viruses visualized with FITC-conjugated anti-pig IgG antibody (green); DAPI, the nuclei were stained by DAPI reagent (blue). All images were visualized and captured at ×200 magnification. DAPI: 4′,6-diamidino-2-phenylindole; FITC: fluorescein isothiocyanate; hTERT: human telomerase reverse transcriptase; IgG: immunoglobulin G; MDCK: Madin-Darby Canine Kidney; MOI: multiplicity of infection; PBEC: porcine bronchial epithelial cell; PCV2: porcine circovirus type 2; PK15: Porcine Kidney 15; SIV: swine influenza virus [Color figure can be viewed at wileyonlinelibrary.com]
4 DISCUSSION
Appropriate cultured cell lines are important and are indispensable to examine the molecular and cellular mechanism of pathogenesis of different hosts. In the study of porcine respiratory pathogens, primary PBECs have been demonstrated to be a valuable in vitro infection model (Blanchard et al., 1992; Meng et al., 2016). In addition, primary PBECs are also used in the study of human pathogens, such as human adenovirus (E. Lam, Ramke, Groos, Warnecke, & Heim, 2011). Here, we described the detailed method for isolation and culture of primary bronchial epithelial cells from a healthy piglet, and we also provided the method for establishment of immortal PBECs for the first time.
To isolate primary PBECs in this study, isolated bronchial tissues were digested for 1 hr via a collagenase I and trypsin digestion method. The short-term digestion of minced porcine bronchial mucosa with microsurgical forceps and scissors yielded large numbers of highly viable and pure epithelial cells. This method had an advantage over isolation techniques used in human and other animal species (H. C. Lam et al., 2011; Ramirez et al., 2004) because weak enzymes and a longer digestion time (16–48 hr) may increase the number of other nonepithelial contaminating cells (Abraham et al., 2011). To promote the growth of PBECs and inhibit the outgrowth of nonepithelial cells, such as smooth muscle cells, culture medium containing epithelial cell regulatory factors was added, and the concentration of FBS was adjusted to 2% after primary PBECs were adherent. In addition, hEGF and retinoic acid were also added after primary PBECs were adherent. EGF and retinoic acid not only play important roles in optimizing air–liquid interface culture growth and differentiation, but may also inform cell culture studies of other species (Bateman, Karasin, & Olsen, 2013; Yoon, Gray, Guzman, Koo, & Nettesheim, 1997).
Immortalized cell lines have numerous advantages over primary cultures, particularly the retention of reasonably constant characteristics for following numerous passages (Miyazawa et al., 2010). However, spontaneous immortalization is a rare event in most somatic cells. Therefore, introduction of foreign genes is required usually through transformation with viral oncogenes, such as SV40 large T antigen, or by using telomerase reverse transcriptase (Miyazawa et al., 2010; Xie et al., 2015), which is generally used to improve the probability of cell immortalization and to eventually obtain immortalized cell lines (Liu, Hatton, Khandelwal, & Sullivan, 2010). Immortalization by viral oncoproteins, such as SV40 large T antigen, to affect host cell-cycle regulation is also a valuable tool but has disadvantages of chromosomal abnormalities, alterations in cell-cycle control compared to primary cells (Moffatt-Jauregui et al., 2013) and an increased risk of tumorigenicity that differs from their untransformed counterparts (Liu et al., 2010). Normal somatic cells, such as epithelial cells, are incapable of indefinite proliferation because their life span is limited by cellular senescence. Previous studies have confirmed that shortened telomeres may be the main cause of cellular senescence (Bodnar et al., 1998). Induction of telomerase activity is a good strategy for reducing cell senescence by preventing telomere shortening (Bodnar et al., 1998; Liu et al., 2010). In this respect, overexpression of hTERT in cells not only prevents telomere shortening but also initiates telomerase activation and extends the life span of cells (Li et al., 2012; Su et al., 2013; Uebing-Czipura et al., 2008). Several studies have obtained immortalized cells through the hTERT overexpression method, including an equine bronchial epithelial cell line (Abraham et al., 2011), a bovine type II alveolar epithelial cell line (Su et al., 2013), a goat mammary epithelial cell line (He et al., 2009), a canine bronchiolar epithelial cell line (Xie et al., 2015), a porcine intestinal epithelial cell line (Wang et al., 2014), and a porcine luteal cell line (L. Zhang et al., 2017).
In the current study, a stable hTERT-PBEC line transfected with hTERT was established, and the morphological and functional characteristics of this cell line were analyzed. Cytokeratins play a critical role in tissue specialization and cell differentiation, functioning to maintain the overall structural integrity of epithelial cells (Calaf & Roy, 2008; Xie et al., 2015). The expression of cytokeratin is considered as the marker of epithelial cells (Schierack et al., 2006; Wang et al., 2014), and cytokeratin 18 immunostaining is used for the identification of fully differentiated epithelial cells (Randell, Walstad, Schwab, Grubb, & Yankaskas, 2001). Here, cytokeratin 18 immunostaining was detected in most of the primary PBECs and hTERT-PBECs, which demonstrated that these cells were epithelial in origin. The analysis of morphological characteristics also showed no obvious differences between primary PBECs and hTERT-PBECs. However, immortal hTERT-PBECs, even after passage 60, also displayed enhanced proliferation capacity compared with primary PBECs. Moreover, consistent with previous reports (He et al., 2009; Su et al., 2013; Wang et al., 2014; Xie et al., 2015), cell cycle analysis by FCM demonstrated that the S phase was increased in hTERT-PBECs. Therefore, these results indicated that the proliferation capacity of hTERT-PBECs is increased compared to primary PBECs.
Previous studies have demonstrated that ectopic expression of hTERT in telomerase-negative normal cells is sufficient to induce telomerase activity and has been used to immortalize the majority of normal and tumor cell types (He et al., 2009; Hong et al., 2007). Here, the overexpression of hTERT in hTERT-PBECs was demonstrated by RT-PCR, western blot analysis and TRAP analysis. The telomerase activity in hTERT-PBECs was significantly higher than that in primary PBECs, indicating that overexpression of hTERT led to the increased telomerase activity in hTERT-PBECs. As expected, immortal hTERT-PBECs had a diploid karyotype and a modal chromosome number of 38, which was consistent with karyotype samples isolated from the male piglet. In addition, the relative quantitative RT-PCR data showed that five genes involved in the cell cycle were highly expressed in hTERT-PBECs compared to primary PBECs, with an exception for sox17. The results may explain why the immortalized cell line demonstrated extended proliferation because some previous studies have revealed that high expression of genes involved in cell cycle control is common for immortalized cells (H. L. Chen et al., 2013; Ha Thi et al., 2014; Klajic et al., 2014; Tompkins et al., 2011). For other genes related to innate immunity and oxidative stress response, similar expression levels were found in hTERT-PBECs and primary PBECs. Moreover, the immortalized hTERT-PBECs were not transformed and did not cause tumors, suggesting that they can be safely used for future studies.
Previous studies demonstrated that interferon responses were induced after porcine airway epithelial cells infected by SIV (Krishna et al., 2015) and differentiated swine airway epithelial cell cultures could be used for influenza A virus infection and replication (Bateman et al., 2013), suggesting that our immortalized hTERT-PBECs may be an ideal cell model for SIV infection. In addition, it is well known that PK15 is the major in vitro cell line suitable to PCV2 infection (Huang et al., 2017; P. Zhang et al., 2018). Recent studies reported that except from PK15 cells, infection of PCV2 in intestinal porcine epithelial cell line (Yan, Zhu, & Yang, 2014) and porcine iliacartery endothelial cells (Yang et al., 2017) could also be detected and observed, suggesting that epithelial cells may also be the target cells of PCV2. Thus, in the current study, SIV and PCV2 were finally selected to perform the indirect immunofluorescence assay to test the susceptibility of immortalized hTERT-PBECs to porcine respiratory viruses. As expected, green fluorescent signals were observed in immortalized hTERT-PBECs infected with both SIV and PCV2. Thus, our established hTERT-PBECs not only retained the morphological and functional features of primary PBECs, but susceptible to porcine respiratory viruses as well, and they may serve as an in vitro infection model.
In summary, we provided detailed protocols for bronchus isolation from piglets and in vitro expansion of primary PBECs. Furthermore, we established the immortalized hTERT-PBEC line, which retains the morphological and functional features of primary PBECs. The establishment of immortal hTERT-PBECs is of great importance as an in vitro model for pathogenicity studies of porcine respiratory pathogens.
ACKNOWLEDGMENTS
This study was supported by the National Natural and Science Foundation of China (L. H., 31400164; Y. Y., 31700158; H. W., 31700157), and the Natural Sciences Foundation of Jiangsu Province (Recipient: Xing Xie, BK20180596; Recipient: Maoda Pang, BK20170600; Z. Z. BK20160583; L. H. BK20140754).
CONFLICTS OF INTERESTS
The authors declare that they have no conflicts of interest.
AUTHORS’ CONTRIBUTIONS
X. X. carried out most of the experiments described in the manuscript and wrote the article; Y. G. , M. D. P., and G. Q. S. participated in the nude mice analysis; L. Z. and B. B. L. were involved in soft agar assay. Q. X. helped with the karyotype analysis; H. Y. W., Y. Y. F., Y. F. Y., R. C., M. W., Z. Z. Z., L. Z. H., Q. Y. X., and M. J. L. were involved in cell cycle analysis, histopathological analysis, and interpretation of data. Z. X. F. conceived the study and contributed in its design and coordination. All authors read and approved the final manuscript.