Are machine learning based methods suited to address complex biological problems? Lessons from CAGI-5 challenges
Abstract
In silico approaches are routinely adopted to predict the effects of genetic variants and their relation to diseases. The critical assessment of genome interpretation (CAGI) has established a common framework for the assessment of available predictors of variant effects on specific problems and our group has been an active participant of CAGI since its first edition. In this paper, we summarize our experience and lessons learned from the last edition of the experiment (CAGI-5). In particular, we analyze prediction performances of our tools on five CAGI-5 selected challenges grouped into three different categories: prediction of variant effects on protein stability, prediction of variant pathogenicity, and prediction of complex functional effects. For each challenge, we analyze in detail the performance of our tools, highlighting their potentialities and drawbacks. The aim is to better define the application boundaries of each tool.
1 INTRODUCTION
Computational tools for predicting the effects of genetic variants are of invaluable importance for complementing experimental approaches in the dissection of the complexity underlying many human diseases. The development of tools for the prediction of variant effects is nowadays a major line of research in bioinformatics and, therefore, many different methods have been described in the past few years (Niroula & Vihinen, 2016). One major issue concerns the ability to effectively assess the prediction performances of available tools to highlight potentialities and drawbacks of each method with respect to different variant-effect-related prediction tasks.
The critical assessment of genome interpretation (CAGI) is a community-wide, international experiment aiming at assessing different methods and approaches for predicting and interpreting the effects of genetic variants. The CAGI is a periodic experiment (typically ran every 2 years), which has reached its fifth edition. The first one has been carried out in 2010 and the last one, the CAGI-5, took place in 2018 and consisted of 14 different prediction challenges covering a wide spectrum of biological problems related to variant effect prediction. Over the years, the CAGI experiment has been significantly contributing to the field, acting as a major driver for testing novel methods and stirring new ideas for variant effect prediction and interpretation.
We have been active participants of the CAGI experiment since its first edition. Indeed, our research activity focuses on the development of tools for genetic variant interpretation, for relating variants and diseases, (Calabrese, Capriotti, Fariselli, Martelli, & Casadio, 2009; Capriotti, Calabrese, & Casadio, 2006; Casadio, Vassura, Tiwari, Fariselli, & Martelli, 2011), and for evaluating the impact of variations on protein stability, (Capriotti, Fariselli, Rossi, & Casadio, 2008; Casadio et al., 2011; Fariselli, Martelli, Savojardo, & Casadio, 2015; Savojardo, Fariselli, Martelli, & Casadio, 2016).
In this paper, we analyse the results obtained by our group on a selection of five different CAGI-5 prediction challenges involving the following genes: FXN (frataxin), TPMT-PTEN (thiopurine S-methyltransferase and phosphatase and tensin homolog), CHEK2 (checkpoint kinase 2), PCM1 (pericentriolar material 1), and GAA (acid α-glucosidase). We classified these challenges into three different categories on the basis of the nature of the underlying prediction task. In particular, we have challenges requiring assessing the impact of variations on protein stability (FXN and TPMT-PTEN), challenges asking to predict variant pathogenicity (CHEK2) and complex challenges requiring to assess different types of functional effects not directly related to the two above categories (PCM1 and GAA). Here, the aim is to summarize our experience on CAGI-5 to highlight pros and cons of our approaches for the different challenge categories, as well as trying to define general guidelines for the proper selection of tools when addressing complex prediction tasks.
2 METHODS
2.1 SNPs&GO: A predictor for annotating the pathogenicity of a protein variation
SNPs&GO (Calabrese et al., 2009) is a method based on support vector machines for predicting the probability of a single amino acid variation (SAV) to be pathogenic. The method elaborates information extracted from protein sequence, multiple sequence alignment (MSA) of similar proteins and protein function. Sequence features include the SAV type and the composition of the environment around the variant site. Features derived from MSA include the frequencies of wild-type and variant residues, the conservation index and the number of aligned sequences. Functional information is encoded by means of a protein-specific (SAV independent) descriptor derived from the distribution of Gene Ontology (GO) annotations in the UniProtKB database. In the preprocessing phase, for each GO term, the frequencies of association to proteins carrying pathogenic and neutral SAVs are estimated and the corresponding log-odd value (LGO) is computed. Then, in the prediction phase, a single descriptor is computed by summing the LGO values of the GO term associated to the input protein (see Calabrese et al., 2009 for details).
SNPs&GO is available as web server at https://snps-and-go.biocomp.unibo.it/snps-and-go/.
2.2 Impact of non-synonymous mutations on protein stability (INPS) and INPS-3D predictors
INPS (Fariselli et al., 2015) is a method for predicting the impact of SAVs on protein stability starting from protein sequence. In particular, it estimates the difference of Gibbs free energy change (ΔG) between wild-type and variant proteins (ΔΔG). INPS adopts a support vector regression approach trained on seven features, six of which are extracted from sequence and one from a hidden Markov model (HMM), whose parameters are estimated from the MSA of chains sharing similarity with the input protein. The features derived from sequence include (a) the BLOSUM62 score corresponding to the substitution from wild-type to variant residues, (b) the Dayhoff mutability index of the wild-type residue, (c,d) the molecular weights and (e,f) Kyte-Doolittle hydrophobicity values of wild-type and of variant residues, respectively. The seventh feature stems from the difference of the HMM-computed Viterbi scores of wild-type and variant sequences.
When necessary, we used INPS-3D (Savojardo et al., 2016), which extends INPS by including, when available, features extracted from the protein 3D structure. These consider the relative solvent accessibility as computed by DSSP (Kabsch & Sander, 1983) and the local energy change upon variation, estimated as the difference between average pairwise residue-contact potential (Bastolla, Farwer, Knapp, & Vendruscolo, 2001) of wild-type and variant proteins.
INPS and INPS-3D are both available through the INPS-MD web server at https://inpsmd.biocomp.unibo.it.
2.3 Pathogenicity and perturbation: Pd and Pp indexes
The disease and perturbation probability indexes (Pd and Pp; Casadio et al., 2011) associate each SAV type (i.e., wild-type and variant residue pair) to the probability of being disease-related (Pd) and of perturbing the protein stability (Pp), respectively. The probability indexes were statistically derived from a data set of 17,170 SAVs in 5,305 proteins retrieved from data available at UniProtKB (release 2010_04), dbSNP (build 132), OMIM and ProTherm (Kumar et al., 2006). The databases include variations related to disease, neutral variants as well as effects of variants on protein thermodynamic stability (Casadio et al., 2011). Only SAVs deriving from single-nucleotide variations (SNPs) are considered. Moreover, SAV types lacking associated thermodynamic data in ProtTherm were filtered out. As a result, Pd and Pp are available for 141 SAV types (Casadio et al., 2011).
2.4 CAGI-5 challenges
Our research group participated in several challenges of the fifth edition of the CAGI (CAGI-5), which took place in 2018. Our submissions were based (directly or indirectly) on previously developed tools for assessing whether protein variations are related to disease, including SNPs&GO and the disease probability indexes (Pd), and tools for the prediction of the impact of protein variants on protein stability like INPS/INPS-3D predictors and the perturbation probability index (Pp).
-
Challenges related to prediction of the effect of variations on protein stability, measured both directly (Frataxin) or indirectly (TPMT-PTEN) using different experimental methods.
-
Challenges related to the evaluation of the pathogenicity of protein variations, as assessed directly in humans (CHEK2).
-
Challenges that require to address complex problems of different nature, including the evaluation of functional effects of variations as assessed on model organisms different from humans (PCM1) or functional effects not directly related to protein stability and/or disease onset (GAA).
In the following, we will describe our approaches for each of the challenges.
2.4.1 Frataxin challenge
For the CAGI-5 Frataxin challenge, participants were provided with a data set comprising eight somatic SAVs of the frataxin protein (FXN), extracted from the Catalog of Somatic Mutations in Cancer (COSMIC) database (Tate et al., 2018) and already known as involved in neoplastic disease and/or cancer. Predictors were asked to submit, for each SAV in the data set, ΔΔG values in kcal/mol, namely the difference of unfolding free energies (ΔG) between mutant and wild-type proteins, extrapolated at concentration zero of denaturant. Experimental ΔΔGs were obtained at the Sapienza University, Rome, Italy (Petrosino et al., 2019).
We tackled the Frataxin challenge running the INPS-3D predictor directly on the available reference PDB structure (1EKG) and obtaining a ΔΔG value for each SAV. Raw INPS-3D predictions were then submitted without any post-processing.
2.4.2 TPMT-PTEN challenge
In the CAGI-5 TPMT-PTEN challenge, predictors were asked to estimate the impact on protein stability of a large panel of SAVs of human thiopurine S-methyltransferase (TPMT) and phosphatase and tensin homolog (PTEN) proteins. Experimental stability scores were assessed by data providers using a multiplexed variant stability profiling (VSP) assay, which uses a fluorescent reporter system to measure the steady-state abundance of missense protein variants (Yen, Xu, Chou, Zhao, & Elledge, 2008). Submitted predictions needed to be scaled in the range [0, +∞], where a value equal to 0 means that the variant is totally unstable, 1 means wild-type stability (neutral) and >1 means stability greater than the wild type.
We predicted the impact on protein stability using both INPS and INPS-3D predictors. In particular, we first mapped SAVs on available 3D structures from PDB and we predicted ΔΔGs with INPS-3D. All remaining SAVs that could not be mapped on 3D structures, were predicted using INPS on the PTEN and TPMT sequences available at UniProtKB.
ΔΔG values predicted by either INPS or INPS-3D were calibrated and rescaled in the required range using data from a functional characterization study of Salavaggione et al., (2005). In this study, functional effects were experimentally evaluated for 11 TPMT SAVs (not included in the challenge data set). We used this experimental evidence for estimating a linear model to map INPS and INPS-3D ΔΔGs onto the requested range (1 = wild type, 0 = totally destabilizing, >1 = stabilizing). The same calibration procedure was applied to both proteins. For sake of comparison, we complemented our predictions including the protein stability perturbation probability index (Pp).
2.4.3 CHEK2 challenge
The CAGI-5 CHEK2 challenge focus on variants of the human checkpoint kinase 2 (CHEK2), which is involved in breast cancer. Data provided include a panel of 34 SAVs obtained from targeted resequencing study on 1,000 Latina breast cancer cases and 1,000 ancestry-matched controls. Predictors were asked to provide the probability pcase for a variant to occur in a case. A pcase > .5 means that the variation is pathogenic, a pcase = .5 means that the variation is neutral (occurring with the same frequency in both populations) while a value below .5 indicate that the variation is protective.
For this challenge, we used both the disease probability index (Pd) and SNPs&GO to assess pathogenicity of each variant. Furthermore, we complemented the above approaches with methods for assessing protein perturbation, including INPS and the perturbation probability index (Pp).
All the methods did not provide information about protective variants, hence predictions are limited to pcase ≥ 0.5. When Pd or Pp are used, we predicted a pcase = 1 for all variations having Pd (Pp) 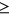 0.8 and a pcase = 0.5 for all variations with Pd (Pp) ≤ 0.4, while values 0.4 < Pd (Pp) < 0.8 were linearly rescaled in the range [0.5,1].
0.8 and a pcase = 0.5 for all variations with Pd (Pp) ≤ 0.4, while values 0.4 < Pd (Pp) < 0.8 were linearly rescaled in the range [0.5,1].
From SNPs&GO output, we derived a pcase in the range [0.5,1] using class predictions (C, neutral or disease) and reliability indexes (RI, from 0 to 10). In particular, we linearly mapped SNPs&GO output to [0.5,1] such that a prediction (C = neutral, RI = 8) corresponds to pcase = 0.5 and a prediction (C = disease, RI = 8) corresponds to pcase = 1. We set the maximum RI to 8 because this is the maximum value found in this particular set of predictions.
INPS ΔΔG output was rescaled in the range [0.5,1] such that pcase = 1 if |ΔΔG| ≥ 1 kcal/mol, pcase = 0.5 if 0.0 kcal/mol ≤ |ΔΔG| < 0.5 kcal/mol while any 0.5 kcal/mol < |ΔΔG| < 1.0 kcal/mol was identically mapped in the range [0.5,1].
2.4.4 PCM1 challenge
The CAGI-5 PCM1 challenge required to predict the effect of a set of SAVs on zebrafish brain development. In particular, a panel of 38 variants within the pericentriolar material 1 (PCM1) gene was assayed on a zebrafish model to determine their impact on the volume of the posterior ventricle area. SAVs were then classified in three different categories: benign (having no impact on zebrafish brain formation), pathogenic (completely disrupting brain formation), and hypomorphic, characterized by a partial loss of function.
Our submission was based on predictions obtained using the disease probability index (Pd), which assigns to each SAV the probability of being associated with the disease. According to the Pd values, variants were assigned with a functional effect, considering a SAV as pathogenic when Pd ≥ 0.6, hypomorphic when 0.4 < Pd < 0.6 and benign if Pd ≤ 0.4.
We complemented the above approach with predictions based on the assessment of the impact of SAVs on protein stability. In particular, we adopted the perturbation probability index (Pp) and the INPS predictor (using the entry UniProtKB Q15154). Similarly to Pd, thresholds on values of Pp were defined for assigning variant function effects (the same thresholds, 0.6 and 0.4, were adopted). INPS ΔΔG predictions were mapped to three classes according to the following scheme: pathogenic if |ΔΔG| ≥ 1 kcal/mol, hypomorphic if 0.5 kcal/mol ≤ |ΔΔG| < 1 kcal/mol and benign if |ΔΔG| < 0.5 kcal/mol.
2.4.5 GAA challenge
The CAGI-5 GAA challenge focused on predicting the effect of naturally occurring variations on enzymatic activity of the human glucosidase α acid (GAA). Experimental enzymatic activity was assessed by data providers (BioMarin Pharmaceutical) for 356 novel missense mutations extracted from the ExAC data set. Plasmids containing complementary DNAs (cDNAs) encoding each of the mutant proteins were transfected into an immortalized Pompe patient fibroblast cell line, with no GAA activity. After 72 hr, cells were lysed, and GAA activity in the lysate was assessed with a fluorogenic substrate. Participants were asked to submit, for each variant in the panel, a numeric value v ≥ 0 representing relative enzymatic activity with respect to wild type: v = 0 indicates no activity, v = 1 wild-type activity, v > 1 increased activity with respect to wild type.
Our submission for this challenge was based on SNPs&GO whose output was linearly rescaled in the range [0,1]. Here we included predictions obtained with the disease and perturbation probability indexes (Pd and Pp), and INPS. Pd and Pp were directly used without any preprocessing. INPS ΔΔG outputs were linearly remapped in the range [0,1].
2.5 Scoring the predictions
To score the prediction, we divided the five CAGI challenges into two categories: Frataxin, TPMT-PTEN, and GAA are regression tasks, whereas CHEK2 and PCM1 are binary classification problems.
-
Pearson correlation coefficient (⍴)
-
Spearman rank correlation coefficient (rs)
-
Kendall Tau rank correlation coefficient (τ)
-
Root mean square error (RMSE)
-
Mean absolute error (MAE)
-
Sensitivity (SEN)
-
Specificity (SPE)
-
Positive predictive value (PPV)
-
Negative predictive value (NPV)
-
Accuracy (ACC)
-
Matthews correlation coefficient (MCC)
-
F1 measure (F1)
3 RESULTS
3.1 Prediction of SAV effect on protein stability: Frataxin and TPMT-PTEN challenges
3.1.1 Frataxin challenge results
As detailed in Section 2, the Frataxin challenge required to submit ΔΔG predictions for eight SAVs of the human frataxin protein (FXN). We evaluated the performance of our INPS-3D predictor with the regression analysis (Table 1). A comparison of experimental and predicted ΔΔG is shown (Figure 1). When INPS-3D is scored as binary classifier, the eight variants are split into in two subsets, corresponding to destabilizing and nondestabilizing SAVs. In particular, adopting a threshold of −1 kcal/mol on experimental ΔΔG, five out of eight variants are destabilizing. The same threshold is applied to INPS-3D predictions. Table 1 lists classification scoring indexes.
| Methods | Regressiona | Classificationa | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
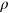 |
rs | τ | RMSE | MAE | SEN | SPE | PPV | NPV | ACC | MCC | F1 | |
| INPS-3D | 0.71 | 0.62 | 0.43 | 3.05 | 2.24 | 0.6 | 1.0 | 1.0 | 0.6 | 0.75 | 0.6 | 0.75 |
- Abbreviations: ACC, accuracy; INPS, impact of nonsynonymous mutations on protein stability; MAE, mean absolute error; MCC, Matthews correlation coefficient; NPV, negative predictive values; PPV, positive predictive value; RMSE, root mean square error; SEN, sensitivity; SPE, specificity.
- a For scoring indexes definition see the scoring the predictions paragraph in Section 2.

Predicted vs experimental ΔΔG values for the eight variants of the human frataxin protein (FXN). Predictions were obtained using INPS-3D
Results indicate that INPS-3D performs very well on the task, achieving very high performances in both regression and classification schemes. According to the official CAGI-5 assessment, INPS-3D is among the top-performing methods participating to this challenge.
Our method fails on predicting the single SAV p.Trp173Cys (Figure 1). This variant is associated to a very low experimental ΔΔG value of −9.5 kcal/mol. As stated during the official assessment, the protein variant p.Trp173Cys corresponds to a clear unfolded state of the protein as experimentally determined (Petrosino et al., 2019; Savojardo et al., 2019). For this reason, the data providers assigned to the variant an arbitrary ΔG of 0 kcal/mol and, as a consequence, a very low ΔΔG value. Removing the outlier from the evaluation, RMSE and MAE decrease down to 1.64 and 1.48 kcal/mol, respectively. These values are much lower than the ones reported in Table 1 and closer to performances reported by INPS-3D in other benchmarks (Savojardo et al., 2016).
Overall, we can conclude that the performance of INPS-3D in the challenge is similar to the one already described in previous papers when predicting experimentally determined ΔΔG values, as expected, given that our method was trained on the same type of data (Savojardo et al., 2016).
INPS-3D predictions for FXN SAVs are reported in Table S1.
3.1.2 TPMT-PTEN challenge results
For the TPMT-PTEN challenge, a set of SAVs of human thiopurine S-methyltransferase (TPMT) and phosphatase and tensin homolog (PTEN) were provided to participants. The task was to compute the effect of each SAV on protein stability (i.e., a numeric score > 0), representing relative stability with respect to wild type (see Section 2 for details). During challenge evaluation, after excluding SAVs with negative experimental scores as well as stop-gain variants, assessors retained 7,473 SAVs, 3,860, and 3,613 on PTEN and TPMT, respectively. This data set was predicted adopting a combined approach based on INPS and INPS-3D (the method used for our official submission) as well as on the perturbation probability index (Pp). Table 2 lists results of the regression analysis. Since Pp is only defined for 141 SAV types, we were able to provide predictions for a subset of 2,982 out of 7,473 SAVs in the data set (1,556 from PTEN and 1,426 from TPMT). In Figure 2 results obtained with INPS + INPS-3D are shown adopting a scatter plot between experimental and predicted stability scores.
| Methods | Data set | 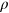 |
rs | τ |
|---|---|---|---|---|
| INPS + INPS-3D | PTEN | 0.50 | 0.44 | 0.30 |
| INPS + INPS-3D | TPMT | 0.39 | 0.37 | 0.25 |
| INPS + INPS-3D | TPMT + PTEN | 0.44 | 0.39 | 0.27 |
| Pp | PTEN* | 0.18 | 0.16 | 0.11 |
| Pp | TPMT** | 0.17 | 0.16 | 0.11 |
| Pp | TPMT + PTEN*** | 0.18 | 0.16 | 0.11 |
- Abbreviations: INPS, impact of nonsynonymous mutations on protein stability; PTEN, phosphatase and tensin homolog; TPMT, thiopurine S-methyltransferase.
- * Predictions obtained on the subset of 1,556 PTEN SAVs for which Pp is defined.
- ** Predictions obtained on the subset of 1,426 TPMT SAVs for which Pp is defined.
- *** Predictions obtained on the subset of 2,982 PTEN + TPMT SAVs for which Pp is defined.

Predicted vs experimental stability scores for the 7,473 variants of human TPMT (3,613 variants) and PTEN (3,860 variants). Predictions obtained with INPS + INPS-3D are shown individually for PTEN (a) and TPMT (b) variants and for the whole data set (c). INPS, impact of nonsynonymous mutations on protein stability; PTEN, phosphatase and tensin homolog; TPMT, thiopurine S-methyltransferase
When comparing the two approaches, it appears that INPS + INPS-3D (based on machine learning) outperforms Pp, which is a simple statistical approach. ΔΔG predictions obtained with INPS + INPS-3D (which were rescaled linearly in the required range) significantly correlate with experimental values, achieving, on the whole data set of SAVs, Pearson's and Spearman's correlation coefficients of 0.44 and 0.39, respectively.
Our strategies show different performances on the two proteins, with correlations that are lower for TPTM and higher for PTEN. Overall, our INPS + INPS-3D submissions are in the top 50% among challenge participants as highlighted in the assessment.
Comparing results of Frataxin and TPMT-PTEN challenges, it is worth noting that, using the same prediction approach, we achieved very different levels of performance (cf. correlation coefficients in Tables 1 and 2). Moreover, prediction performance of INPS and INPS-3D in TPMT-PTEN are far below those previously achieved with the same methods in several benchmark datasets (Fariselli et al., 2015; Savojardo et al., 2016). A possible interpretation of the results is that when the methods are adopted to predict experimental thermodynamic stability data (ΔΔG), they perform better since they have been trained on the same type of data. As soon as the data type differs (for TPMT-PTEN, the impact of SAVs on protein stability was measured using a large-scale multiplexed VSP assay), the performance decreases.
Predictions for TPMT-PTEN SAVs are reported in Table S2.
3.2 Prediction of SAV pathogenicity: CHEK2 challenge
For the CHEK2 challenge participants were asked to provide predictions of pathogenicity for 34 SAVs of the human checkpoint kinase 2 (CHEK2). Here, we evaluated performances of methods devised to predict the relation of SAVs with diseases, such as Pd and SNPs&GO, as well as methods devised to predict impact of SAVs on protein stability like Pp and INPS. Outputs of all methods were rescaled so as to provide a numerical value, referred to as pcase, which represents the probability of each SAV to be pathogenic (see Section 2 for details). Binary classification of SAVs in the data set was obtained by applying a threshold on the pcase value. Table 3 lists the results obtained with pcase threshold set to 0.75.
| Methods | SEN | SPE | PPV | NPV | ACC | MCC | F1 |
|---|---|---|---|---|---|---|---|
| Pd | 0.52 | 0.69 | 0.73 | 0.47 | 0.59 | 0.21 | 0.61 |
| SNPs&GO | 0.71 | 0.85 | 0.88 | 0.65 | 0.76 | 0.54 | 0.79 |
| Pp | 0.19 | 0.77 | 0.57 | 0.37 | 0.41 | −0.05 | 0.29 |
| INPS | 0.52 | 0.69 | 0.73 | 0.47 | 0.59 | 0.21 | 0.61 |
- Abbreviations: ACC, accuracy; GO, Gene Ontology; INPS, impact of nonsynonymous mutations on protein stability; MCC, Matthews correlation coefficient; NPV, negative predictive values; PPV, positive predictive value; SEN, sensitivity; SNP, single nucleotide polymorphism; SPE, specificity.
- Note: Classification scoring indexes were computed setting pcase threshold to 0.75 for identifying pathogenic variants.
Among the different approaches evaluated, SNPs&GO is the best-performing one, reporting an MCC value of 0.54. The pattern of mispredictions of SNPs&GO in this challenge is very similar to what already assessed for the predictor in much larger datasets (Calabrese et al., 2009). As a rule of thumb, SNPs&GO tends to more precise than sensitive (i.e., it is characterized by a high PPV and a lower sensitivity). The same behavior can be observed in the CHEK2 challenge, where SEN and PPVs are 0.71 and 0.88, respectively. During the official challenge assessment, SNPs&GO was scored as the top-performing method.
Comparing performances of the different methods, it is evident that those that directly predict SAV pathogenicity (SNPs&GO and Pd) tend to outperform those that are instead devised to predict impact of SAV on protein stability (INPS and Pp). This suggests that methods implemented for predicting impact of SAVs on thermodynamic stability can be helpful in assessing SAV pathogenicity but, in many cases, they are not sufficient for obtaining accurate predictions.
Predictions for CHEK2 SAVs are reported in Table S3.
3.3 Complex prediction challenges: PCM1 and GAA
3.3.1 PCM1 challenge results
The PCM1 challenge required to classify a set of 38 SAVs of the pericentriolar material 1 (PCM1) gene into three different classes (pathogenic, hypomorphic, and benign) according to the estimated impact on brain development as measured on a zebrafish model. Following the same approach adopted during the challenge assessment, predictions were scored using a binary classification scheme, which collects into a single class pathogenic and hypomorphic SAVs and evaluates the ability of methods in discriminating them from benign SAVs.
In Table 4, we report classification results obtained in this task with Pd, Pp, and INPS.
| Methods | SEN | SPE | PPV | NPV | ACC | MCC | F1 |
|---|---|---|---|---|---|---|---|
| Pd | 0.86 | 0.00 | 0.54 | 0.00 | 0.50 | -0.25 | 0.67 |
| Pp | 0.82 | 0.31 | 0.62 | 0.56 | 0.61 | 0.15 | 0.71 |
| INPS | 0.64 | 0.50 | 0.64 | 0.50 | 0.58 | 0.14 | 0.64 |
- Abbreviations: ACC, accuracy; INPS, impact of nonsynonymous mutations on protein stability; MCC, Matthews correlation coefficient; NPV, negative predictive values; PPV, positive predictive value; SEN, sensitivity; SPE, specificity.
Results highlight that all methods evaluated are essentially failing in this challenge, reporting MCC scores that are close to randomness and, in some case, even negative. Interestingly, our official submission for this challenge (the one based on Pd), scoring with an MCC of −0.25, was globally ranked as the third top-performing among all participating methods (global ranks were computed averaging individual ranks computed for each scoring index). Our conclusion would be that our predictors are not suited to capture the complexity of the biological process leading to the different results.
Predictions for PCM1 SAVs are reported in Table S4.
3.3.2 GAA challenge results
The GAA challenge requires to predict the impact on enzymatic activity of 356 SAVs of the human acid α-glucosidase (GAA) protein. In this task, we compared Pd, SNPs&GO, Pp, and INPS. Results of regression analyses are reported in Figure 3 and Table 5.

Predicted vs experimental enzymatic activity scores for 356 variants of human GAA protein. Predictions were generated with Pd (a), SNPs&GO (b), Pp (c), and INPS (d). GAA, glucosidase α acid; GO, Gene Ontology; INPS, impact of nonsynonymous mutations on protein stability; SNP, single nucleotide polymorphism
| Methods |  |
rs | τ | RMSE | MAE |
|---|---|---|---|---|---|
| Pd | 0.17 | 0.18 | 0.12 | 0.36 | 0.30 |
| SNPs&GO | 0.28 | 0.28 | 0.19 | 0.42 | 0.35 |
| Pp | 0.10 | 0.09 | 0.06 | 0.37 | 0.32 |
| INPS | 0.16 | 0.14 | 0.09 | 0.97 | 0.80 |
- Abbreviations: GO, Gene Ontology; INPS, impact of nonsynonymous mutations on protein stability; MAE, mean absolute error; RMSE, root mean square error; SNP, single nucleotide polymorphism.
In our tests, pathogenicity predictors (Pd and SNPs&GO) significantly outperform stability ones (Pp and INPS). However, our submission (based on SNPs&GO), is characterized by a Pearson's correlation value of 0.28 and interestingly ranked among the top-scoring ones (the fourth in terms of individual submissions and the second in terms of research groups). Our interpretation is that again the complexity of the detection system hampers direct predictions that can be addressed by our tools.
Predictions for GAA SAVs are reported in Table S4.
4 CONCLUSION
In this paper we summarized our experience as participants to the fifth edition of CAGI. In particular, we focused on five different challenges which for sake of simplicity, we divided into three different categories: (a) prediction of protein stability perturbation upon variation, (b) prediction of variant pathogenicity, and (c) prediction of complex functional effects. For each challenge, we analyzed the prediction performance of our CAGI official submissions as well as performance of other complementing approaches.
Overall, results on the five challenges here considered confirming the superiority of machine-learning based approaches (SNPs&GO and INPS/INPS-3D) over methods based on basic statistical analyses (Pd and Pp). Our methods perform well when the test set contains data homogeneous to those of the training set. As an example, when predicting SAV effects on protein stability, our methods perform better in the case of Frataxin than in the case of TPMT-PTEN. Indeed, these latter ΔΔG values of TPMT-PTEN variations are not directly measured, as in the case of Frataxin, rather indirectly evaluated from a large-scale multiplexed VSP assay (Yen et al., 2008). Again, when predicting variant pathogenicity of CHEK2, our SNPs&GO is satisfactory performing, given the similarity between the training procedure and the required task. However, when predicting on what we call complex prediction challenges (such as the pathogenicity of PCM1 and GAA variants) even machine-learning approaches fail. Our tools are indeed able to capture the binary classification of simple sets of molecular data directly annotated (Calabrese et al., 2009; Casadio et al., 2011). When classification is derived indirectly with in vivo approaches, possibly it implies complex biological processes, which deserve other models for their simulation.
ACKNOWLEDGMENTS
The CAGI experiment coordination is supported by NIH U41 HG007346 and the CAGI conference by NIH R13 HG006650.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.