

Design, synthesis, and structure–activity relationships of diindolylmethane derivatives as cannabinoid CB2 receptor agonists
Abstract
3,3′-Diindolylmethane (DIM), a natural product-derived compound formed upon ingestion of cruciferous vegetables, was recently described to act as a partial agonist of the anti-inflammatory cannabinoid (CB) receptor subtype CB2. In the present study, we synthesized and evaluated a series of DIM derivatives and determined their affinities for human CB receptor subtypes in radioligand binding studies. Potent compounds were additionally evaluated in functional cAMP accumulation and β-arrestin recruitment assays. Small substituents in the 4-position of both indole rings of DIM were beneficial for high CB2 receptor affinity and efficacy. Di-(4-cyano-1H-indol-3-yl)methane (46, PSB-19837, EC50: cAMP, 0.0144 µM, 95% efficacy compared to the full standard agonist CP55,940; β-arrestin, 0.0149 µM, 67% efficacy) was the most potent CB2 receptor agonist of the present series. Di-(4-bromo-1H-indol-3-yl)methane (44, PSB-19571) showed higher potency in β-arrestin (EC50 0.0450 µM, 61% efficacy) than in cAMP accumulation assays (EC50 0.509 µM, 85% efficacy) while 3-((1H-indol-3-yl)methyl)−4-methyl-1H-indole (149, PSB-18691) displayed a 19-fold bias for the G protein pathway (EC50: cAMP, 0.0652 µM; β-arrestin, 1.08 µM). DIM and its analogs act as allosteric CB2 receptor agonists. These potent CB2 receptor agonists have potential as novel drugs for the treatment of inflammatory diseases.
1 INTRODUCTION
Cannabinoid (CB) receptors belong to the superfamily of G protein-coupled receptors (GPCRs)[1]; the CB1 receptor is the most highly expressed GPCR in the brain.[2] In addition, it is also expressed in peripheral organs such as the lungs, liver, and kidneys.[3] The CB1 receptor modulates neurotransmitter release and has been proposed as a potential drug target for the treatment of pain, neurodegenerative,[4, 5] and metabolic diseases.[6, 7] CB2 receptors are predominantly expressed on immune cells, including macrophages and leukocytes,[8-10] and in organs associated with the immune system, for example, tonsils, spleen, and thymus.[10, 11] Therefore, CB2 receptors are potential therapeutic targets for the treatment of inflammatory diseases.[12] Both CB receptor subtypes couple to Gi/o proteins,[13] and the activation of these receptors results in the inhibition of adenylate cyclase, which leads to reduced intracellular cAMP levels.
CBs that target CB receptors are subdivided into three classes: endocannabinoids, which are naturally produced in the body, phytocannabinoids, derived from plants, and synthetic CBs obtained by chemical synthesis.[12, 14, 15] The main psychoactive component of the herbal drug marijuana, Cannabis sativa, is Δ9-tetrahydrocannabinol (∆9-THC, 1, Figure 1), a balanced partial agonist of CB1 and CB2 receptors.[16, 17] It is used for the therapy of muscle spasms, nausea, and cachexia, and has the potential for a number of further indications.[18, 19] In recent years, a wide range of synthetic CB1 or CB2 receptor agonists has been developed (see examples 2 and 3 in Figure 1), and several approved nonselective CB1/CB2 receptor agonists such as nabiximol, nabilone, and dronabinol are in clinical use. Herbal products containing added synthetic CBs, known as “spice,” have been found on the illicit drug market.[20, 21] Due to their illegal use, many synthetic CBs are included in the list of controlled substances.

It is well known that the psychoactive effects of CBs are mediated by activation of the CB1 receptor expressed in the brain. Selective CB2 receptor ligands are expected to be safer since they are not prone to drug abuse. CB2 receptor agonists have been proposed for the treatment of neurodegenerative and neuroinflammatory diseases, including Alzheimer's, Huntington's, and Parkinson's disease, multiple sclerosis, and amyotrophic lateral sclerosis.[22-25] Moreover, CB2 receptor agonists may be effective in the treatment of irritable bowel syndrome, myocardial infarction, bone disorders, and different types of cancer.[18, 26, 27] However, none of the CB2 receptor ligands studied in clinical trials has thus far received approval as a drug, primarily because of side effects.[28, 29]
Indole represents an important privileged core structure found in many biologically active natural products and synthetic drugs.[11, 20] Indole constitutes an important scaffold among synthetic CBs (see, e.g., 3, JWH-018).[21, 30-32] 3,3′-Diindolylmethane (DIM, 4), a metabolite of indole-3-carbinol, has been reported to exhibit various biological activities, including anticancer effects.[24, 25] In addition to its CB receptor interaction, DIM has been linked to further biochemical targets: It has been shown to activate the arylhydrocarbon receptor (at 30 μM),[26, 27, 30] and the immunostimulatory orphan GPCR GPR84 (at submicromolar concentrations).[33] Furthermore, it was found to block the androgen receptor (at a concentration of ca. 50 μM), and the enzyme histone deacetylase-1 (HDAC-1) at a concentration of 100 μM, which is close to its solubility limit. In a previous study, Yin et al. identified DIM as a CB2 receptor agonist upon screening of a compound library.[34] CB2 receptor interaction and activation by DIM were confirmed in receptor radioligand binding and functional assays including β-arrestin recruitment, [35S]GTPγS binding, and cAMP assays.[34] DIM was reported to display potency in all four assays, with Ki/IC50 values ranging from 0.42 to 1.7 µM. It was found to act as a partial CB2 receptor agonist when compared to the full agonist CP55,940 (2, Figure 1). DIM was also able to bind to the human CB1 receptor (Ki > 4.3 µM) but acted as an antagonist/inverse agonist at that receptor subtype (IC50 11.1 µM).[34] Recently, we developed a synthetic method for the preparation of unsymmetrical 3,3′-DIM derivatives and analogs. Preliminary biological studies of some new DIM derivatives indicated structure-dependent interactions with CB receptors.[35] In the present study, we selected DIM (4) as a lead structure to perform an in-depth study of structure–activity relationships (SARs) at the human CB receptors, mainly with the aim of improving the compound's potency, efficacy, and selectivity for the CB2 receptor subtype. Due to the CB2 receptor selectivity of DIM and its expected allosteric nature, which might result in functional selectivity, this new class of CB receptor agonists appeared to be attractive.
2 RESULTS AND DISCUSSION
2.1 Chemistry
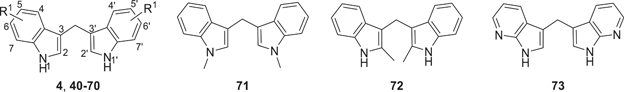
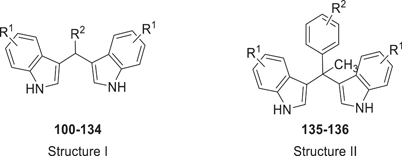
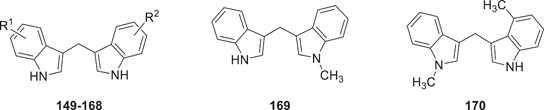
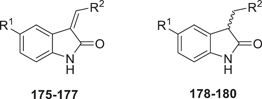
The synthesis of symmetrical DIM derivatives 40–73 (Table 1, Scheme 1) without a substituent on the methylene bridge was performed by reaction of indoles 5–39 with formaldehyde (37% in water) upon microwave irradiation, according to a previously developed optimized method.[33] Symmetrical DIMs (100–136, Table 2, Scheme 2) containing substituents on the bridging methylene group were synthesized by reaction of indoles 5, 7-8, 13–15, 27, 30, and 32 with various aldehydes or ketones (74–99) in water in the presence of concentrated sulfuric acid.[35] Unsymmetrical DIMs 149–170 (Table 3, Scheme 3) were synthesized based on two different procedures: (i) reaction of (3-indolylmethyl)trimethylammonium iodides 137–140 with a range of substituted indole derivatives (6, 8, 10, 13–15, 17, 21, 23, 20, 30, 37, 38, 141, 142, or 143) in water[36] or (ii) decarboxylative coupling of indoles 144–148 with various indolylacetic acid derivatives.[37] The synthesis of 2-oxoindole derivatives was performed according to reported procedures (Table 4, Scheme 4).[33]
 |
|||
| Compounda | R1 | Human CB2 receptor | Human CB1 receptor |
|---|---|---|---|
| Radioligand binding assay | |||
| Ki ± SEM (µM) | Ki ± SEM (µM) | ||
| (or percent inhibition of [3H]CP55,940 at 5 µM) | (or percent inhibition of [3H]CP55,940 at 5 µM) | ||
| [Maximal inhibition (%)] | [Maximal inhibition (%)] | ||
| 4 (DIM) | H | 0.690 ± 0.159 | 5.42 ± 1.00 |
| [98%] | [86%] | ||
| (1.1)a | (4.3)a | ||
| 40 | 4-CH3 | 0.845 ± 0.086 | >5 (4%) |
| [81%] | |||
| 41 | 4-OCH3 | 0.579 ± 0.157 | 5.03 ± 2.29 |
| [94%] | [79%] | ||
| 42 (PSB-16357) | 4-F | 0.279 ± 0.056 | >5 (28%) |
| [99%] | |||
| 43 | 4-Cl | 0.332 ± 0.230 | 0.753 ± 0.048 |
| [93%] | [61%] | ||
| 44 (PSB-19571) | 4-Br | 0.374 ± 0.074 | 7.27 ± 0.45 |
| [100%] | [99%] | ||
| 45 | 4-NO2 | >5 (29%) | >5 (6%) |
| 46 (PSB-19837) | 4-CN | 0.339 ± 0.061 | ≥10 (47%) |
| [99%] | |||
| 47 | 5-CH3 | 2.78 ± 1.36 | >5 (26%) |
| [86%] | |||
| 48 (PSB-16105) | 5-OCH3 | 2.84 ± 1.51 | 5.89 ± 1.28 |
| [83%] | [62%] | ||
| 49 (PSB-15160) | 5-F | 1.17 ± 0.33 | 4.08 ± 0.22 |
| [100%] | [96%] | ||
| 50 | 5-CF3 | 1.98 ± 0.14 | 9.94 ± 3.86 |
| [100%] | [100%] | ||
| 51 | 5-Cl | 0.747 ± 0.067 | >5 (6%) |
| [73%] | |||
| 52 | 5-Br | 1.27 ± 0.23 | >5 (34%) |
| [100%] | |||
| 53 | 5-CN | >5 (30%) | >5 (35%) |
| 54 | 5-NO2 | ≥5 (45%) | >5 (30%) |
| 55 | 5-CO2Me | 2.99 ± 0.03 | >5 (19%) |
| [71%] | |||
| 56 | 5-CHO | 7.25 ± 1.39 | >5 (23%) |
| [71%] | |||
| 57 | 5-CO2H | >5 (34%) | >5 (−17%) |
| 58 | 5-OBn | >5 (37%) | 2.95 ± 0.74 |
| [88%] | |||
| 59 | 6-CH3 | 0.504 ± 0.252 | >5 (30%) |
| [76%] | |||
| 60 | 6-OCH3 | >5 (32%) | >5 (26%) |
| 61 (PSB-16358) | 6-F | 0.985 ± 0.094 | >5 (27%) |
| [90%] | |||
| 62 | 6-Cl | 0.911 ± 0.105 | 0.820 ± 0.385 |
| [84%] | [59%] | ||
| 63 | 6-Br | 3.44 ± 0.56 | 5.28 ± 2.02 |
| [100%] | [100%] | ||
| 64 (PSB-16381) | 7-F | ≥5 (49%) | >5 (18%) |
| 65 | 7-OCH3 | ≥5 (48%) | >5 (30%) |
| 66 | 4-Cl,6-Cl | 0.626 ± 0.219 | 1.68 ± 0.32 |
| [90%] | [68%] | ||
| 67 | 5-F,6-Cl | >5 (36%) | >5 (25%) |
| 68 | 4-F,5-F | 3.04 ± 0.78 | 5.34 ± 1.81 |
| [90%] | [70%] | ||
| 69 (PSB-16586) | 5-F,6-F | ≈5 (59%) | >5 (39%) |
| 70 (PSB-16671) | 5-F,7-F | 1.10 ± 0.19 | 2.64 ± 0.28 |
| [100%] | [100%] | ||
| 71 | See structure above | 3.39 ± 1.14 | >5 (35%) |
| [68%] | |||
| 72 | See structure above | ≥5 (48%) | >5 (45%) |
| 73 | See structure above | >5 (3%) | >5 (20%) |
- a Data were obtained from Yin et al.[34]

 |
||||
| Compound | R1 | R2 | Human CB2 receptor | Human CB1 receptor |
|---|---|---|---|---|
| Radioligand binding assay | ||||
| Ki ± SEM (µM) | Ki ± SEM (µM) | |||
| (or percent inhibition of [3H]CP55,940 at 5 µM) | (or percent inhibition of [3H]CP55,940 at 5 µM) | |||
| [Maximal inhibition (%)] | [Maximal inhibition (%)] | |||
| Structure I | ||||
| 100 | H | Me | 5.74 ± 0.30 | 8.15 ± 2.40 |
| [77%] | [71%] | |||
| 101 | H | Ethyl | 0.804 ± 0.249 | 2.70 ± 1.86 |
| [97%] | [90%] | |||
| 102 | H | Propyl | >5 (33%) | >5 (45%) |
| 103 | H | Butyl | >5 (36%) | >5 (−2%) |
| 104 | H | 4-MePh | 2.55 ± 0.28 | 2.51 ± 0.40 |
| [98%] | [100%] | |||
| 105 | H | 3-MePh | 1.79 ± 0.36 | 3.56 ± 1.47 |
| [79%] | [74%] | |||
| 106 | H | 2-MePh | 4.55 ± 1.52 | 2.98 ± 1.76 |
| [59%] | [58%] | |||
| 107 | H | 4-EthylPh | 1.35 ± 0.56 | 4.44 ± 2.16 |
| [95%] | [100%] | |||
| 108 | H | 4-IsopropylPh | 1.55 ± 0.41 | 0.832 ± 0.281 |
| [85%] | [82%] | |||
| 109 | H | 4-MeOPh | 2.41 ± 0.02 | 0.774 ± 0.169 |
| [73%] | [71%] | |||
| 110 | H | 3-MeOPh | 2.07 ± 0.75 | 3.04 ± 1.16 |
| [96%] | [91%] | |||
| 111 | H | 2-MeOPh | 2.72 ± 2.02 | 1.70 ± 0.41 |
| [59%] | [84%] | |||
| 112 | H | 2,3-Methylenedioxy-Ph | 3.04 ± 0.22 | 1.19 ± 0.102 |
| [84%] | [84%] | |||
| 113 | H | 4-PhenoxyPh | >5 (41%) | 0.402 ± 0.306 |
| [80%] | ||||
| 114 | H | 4-(OH)Ph | 7.13 ± 0.617 | >5 (43%) |
| [60%] | ||||
| 115 | H | 4-ClPh | 3.06 ± 0.90 | 1.24 ± 0.40 |
| [94%] | [92%] | |||
| 116 | H | 4-FPh | >5 (44%) | 3.54 ± 1.49 |
| [88%] | ||||
| 117 | H | 4-NO2Ph | 1.57 ± 0.04 | 0.596 ± 0.240 |
| [80%] | [78%] | |||
| 118 | H | Napth-1-yl | >5 (9%) | >5 (7%) |
| 119 | H | 7-MeO-napth-1-yl | >5 (36%) | 0.983 ± 0.463 |
| [69%] | ||||
| 120 | H | Indol-3-yl | >5 (9%) | >5 (18%) |
| 121 | 4-OMe | Ph | >5 (35%) | >5 (44%) |
| 122 | 4-OMe | 4-MeOPh | >5 (26%) | 0.541 ± 0.173 |
| [88%] | ||||
| 123 | 5-OMe | 4-MePh | >5 (42%) | 0.414 ± 0.260 |
| [68%] | ||||
| 124 | 5-OMe | 4-FPh | >5 (18%) | >5 (34%) |
| 125 | 5-OMe | 4-MeOPh | 2.04 ± 0.37 | 0.176 ± 0.649 |
| [91%] | [83%] | |||
| 126 | 5-Me | Ph | >5 (−8%) | >5 (−31%) |
| 127 | 5-Me | 4-MeOPh | 1.30 ± 0.45 | 2.34 ± 0.172 |
| [79%] | [76%] | |||
| 128 | 5-Me | 4-FPh | 2.18 ± 0.29 | 2.69 ± 0.60 |
| [87%] | [92%] | |||
| 129 | 5-F | Napth-2-yl | 2.35 ± 0.19 | 2.79 ± 0.61 |
| [71%] | [80%] | |||
| 130 | 4-F | Pyridin-4-yl | 5.42 ± 0.22 | 4.96 ± 0.17 |
| [92%] | [82%] | |||
| 131 | 5-F | Pyridin-4-yl | >5 (28%) | >5 (47%) |
| 132 | 6-F | Pyridin-4-yl | >5 (29%) | 5.03 ± 1.40 |
| [79%] | ||||
| 133 | 7-F | Pyridin-4-yl | >5 (14%) | >5 (23%) |
| 134 | 5-Cl | Pyridin-4-yl | >5 (9%) | >5 (9%) |
| Structure II | ||||
| 135 | H | 4-CH3 | 0.674 ± 0.529 | ≥5 (44%) |
| [53%] | ||||
| 136 | H | 4-OCH3 | >5 (35%) | 2.84 ± 0.17 |
| [73%] | ||||

 |
||||
| Compound | R1 | R2 | Human CB2 receptor | Human CB1 receptor |
|---|---|---|---|---|
| Radioligand binding assay | ||||
| Ki ± SEM (µM) | Ki ± SEM (µM) | |||
| (or percent inhibition of [3H]CP55,940 at 5 µM) | (or percent inhibition of [3H]CP55,940 at 5 µM) | |||
| [Maximal inhibition (%)] | [Maximal inhibition (%)] | |||
| 149 (PSB-18691) | H | 4-Me | 0.498 ± 0.176 | >10 (35%) |
| [98%] | ||||
| 150 | H | 4-F | 0.758 ± 0.178 | 6.09 ± 1.78 |
| [98%] | [100%] | |||
| 151 | H | 4-Br | 0.944 ± 0.106 | 31.1 ± 7.4 |
| [99%] | [97%] | |||
| 152 | H | 5-OMe | 2.32 ± 0.71 | >5 (33%) |
| [87%] | ||||
| 153 | H | 5-F | 3.78 ± 0.30 | >5 (20%) |
| [85%] | ||||
| 154 | H | 5-Cl | 1.21 ± 0.05 | 4.40 ± 0.40 |
| [93%] | [91%] | |||
| 155 | H | 5-CO2Me | n.da | >5 (41%) |
| 156 | H | 5-CO2H | >5 (20%) | >5 (−7%) |
| 157 | 4-Cl | 4-Br | 0.237 ± 0.006 | 4.07 ± 1.36 |
| [98%] | [92%] | |||
| 158 | 4-Cl | 4-CH3 | 0.536 ± 0.058 | 7.13 ± 1.68 |
| [96%] | [89%] | |||
| 159 | 5-OMe | 4-F | 1.17 ± 0.14 | >5 (39%) |
| [97%] | ||||
| 160 | 5-OMe | 5-F | 2.44 ± 0.27 | >5 (49%) |
| [88%] | ||||
| 161 | 5-OMe | 7-F | 2.82 ± 0.09 | >5 (31%) |
| [91%] | ||||
| 162 | 5-OMe | 4-F, 5-F | 3.74 ± 0.14 | >5 (36%) |
| [98%] | ||||
| 163 | 5-OMe | 4-F, 6-F | 1.64 ± 0.15 | >5 (44%) |
| [98%] | ||||
| 164 | 5-OMe | 5-Me | 1.20 ± 0.28 | >5 (48%) |
| [95%] | ||||
| 165 | 5-OH | 6-F | 12.0 ± 3.2 | 26.5 ± 12.4 |
| [80%] | [82%] | |||
| 166 | 5-OMe | 6-CF3 | 8.93 ± 1.10 | >5 (44%) |
| [82%] | ||||
| 167 | 5-OBn | 6-F | 1.99 ± 0.39 | 1.40 ± 0.07 |
| [96%] | [100%] | |||
| 168 | 5-OBn | 6-CF3 | 4.12 ± 0.49 | 2.87 ± 0.93 |
| [94%] | [100%] | |||
| 169 | See above | >5 (30%) | >5 (16%) | |
| 170 | See above | 0.716 ± 0.001 | ≈ 10 (53%) | |
| [99%] | ||||
- a n.d, not determined.

 |
||||
| Compound | R1 | R2 | Human CB2 receptor | Human CB1 receptor |
|---|---|---|---|---|
| Radioligand binding assay | ||||
| Ki ± SEM (µM) | Ki ± SEM (µM) | |||
| (or percent inhibition of [3H]CP55,940 at 5 µM) | (or percent inhibition of [3H]CP55,940 at 5 µM) | |||
| [Maximal inhibition (%)] | [Maximal inhibition (%)] | |||
| 175 | H | 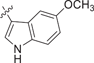 |
>5 (28%) | >5 (38%) |
| 176 | 5-F | 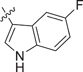 |
3.42 ± 0.22 | 4.72 ± 0.14 |
| [100%] | [100%] | |||
| 177 | 5-F | 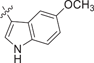 |
>5 (39%) | 9.14 ± 0.87 |
| [100%] | ||||
| 178 | H | 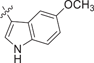 |
>5 (33%) | >5 (33%) |
| 179 | 5-F | 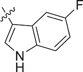 |
>5 (20%) | >5 (33%) |
| 180 | 5-F |  |
>5 (41%) | >5 (16%) |

2-Oxoindole derivatives 171 and 172 were reacted with the appropriate aldehyde (173–174) in ethanol in the presence of piperidine at 65°C to produce α,β-unsaturated indol-2-one derivatives 175–177. These products were reduced by NaBH4 to yield the corresponding saturated indole-2-one derivatives 178–180 (Table 4, Scheme 4).
The structures of the newly synthesized compounds were confirmed by 1H- and 13C-NMR spectra. In addition, HPLC analysis coupled to UV and electrospray ionization mass spectrometry (LC/UV-ESI-MS) was performed, which was also used to determine the purity of the compounds. For all compounds, the purity was above 95%.
2.2 Pharmacology
All compounds were initially evaluated at a concentration of 5 µM for their binding affinity to membrane preparations of Chinese hamster ovary (CHO) cells recombinantly expressing either human CB1 or CB2 receptors. The nonspecific agonist radioligand [3H]CP55,940 was used at a concentration of 0.1 nM for competition binding studies at both receptor subtypes.[21, 30, 38] For compounds that showed displacement of radioligand binding of more than 50%, full concentration-inhibition curves were determined, and Ki values were calculated. Functional assays for the compounds that showed low micromolar Ki values were performed to evaluate their agonistic (or antagonistic) activity. The compounds were evaluated in cAMP accumulation assays according to a described procedure.[21, 30, 39] In addition, agonist-induced β-arrestin recruitment was measured as a G protein-independent pathway using a β-galactosidase complementation assay.[38]
2.2.1 Radioligand binding studies and SARs
A total of 99 DIM derivatives and analogs were evaluated for their binding affinities at the human CB2 receptor. The Ki value of the lead structure DIM (4) in our assay system was 0.690 µM, which is comparable to the reported value of 1.1 µM (see Table 1). SAR analysis revealed that many of the symmetrically substituted indole derivatives displayed higher affinity at the CB2 receptor when compared to the unsymmetrically substituted ones. Substitutions on the methylene bridge of the DIM derivatives were in general not well tolerated, leading to only moderate potency compared to the unsubstituted analogs (all results are collected in Tables 1–4).
The effects of substituents at the 4,4′-position of the indole rings were studied. The following substituents were introduced: 4,4′-dimethyl (40: Ki 0.845 µM), 4,4′-dimethoxy (41: Ki 0.579 µM), 4,4′-difluoro (42: Ki 0.279 µM), 4,4′-dichloro (43: Ki 0.332 µM), 4,4′-dibromo (44: Ki 0.374 µM), 4,4′-dinitro (45: Ki > 5.0 µM), and 4,4′-dicyano (46: Ki 0.339 µM). Substituents in that position could slightly improve potency as compared to the unsubstituted DIM. 4,4′-Difluoro, 4,4′-dichloro, and 4,4′-dicyano substitution was best tolerated.
Next, substituents were introduced at the 5,5′-positions of DIM-yielding compounds 47–58. In general, compounds with 5,5′-substituents on the indole rings showed, in most cases, lower binding affinity compared to the 4,4′-substituted DIM derivatives. The 5,5′-dichloro-DIM (51: Ki 0.747 µM) exhibited the highest affinity among all derivatives of this series. Notably, it displayed only 73% maximum displacement of the radioligand in our assay system. 5,5′-Dimethyl-DIM (47: Ki 2.78 µM), 5,5′-dimethoxy-DIM (48: Ki 2.84 µM), 5,5′-difluoro-DIM (49: Ki 1.17 µM), 5,5′-dibromo-DIM (52: Ki 1.27 µM), 5,5′-ditrifluromethyl-DIM (50: Ki 1.98 µM), and 5,5′-dicarboxylic acid-DIM (57: Ki > 5.0 µM) showed moderate affinities, while 5,5′-diformyl-DIM (56: Ki 7.25 µM), 5,5′-dicyano-DIM (53: Ki > 5.0 µM) and 5,5′-dinitro-DIM (54: Ki > 5.0 µM) were only weakly potent or inactive at a concentration of 5 µM.
Substitution at the 6,6′-positions of DIM did not improve binding affinity (see compounds 59–63 in Table 1). Substituents such as 6,6′-dimethyl (59: Ki 0.524 µM), 6,6′-difluoro (61: Ki 0.985 µM), and 6,6′-dichloro (62: Ki 0.911 µM) were tolerated, showing similar affinity as lead compound 4. Bulkier substituents, namely 6,6′-dimethoxy (60: Ki 5 µM) and 6,6′-dibromo (63: Ki 3.44 µM) reduced the binding affinity. This is probably caused by limited space in the binding site of the CB2 receptor. We next studied 7,7′-substituted DIM derivatives (see Table 1). 7,7′-Difluoro-DIM (64: Ki > 5 µM) and 7,7′-dimethoxy-DIM (65: Ki > 5 µM) derivatives were inactive at 5 µM, indicating that substitutions at the 7,7′-positions are detrimental.
Our next effort was aimed at introducing multiple halogen substituents (compounds 66–70). In general, this modification tended to reduce binding affinity irrespective of the positions of the halogen atoms. Chloro-substitution at the 4- and 6-positions (66: Ki 0.624 µM) was the best combination with equipotent binding affinity to lead compound 4. It was interesting to note that compound 69 was less potent when compared to its 4,4′-dichloro-DIM analog 43 and slightly more potent than its 6,6′-dichloro-DIM derivative (62). Other combinations, as indicated in Table 1 (compounds 67–70), reduced binding affinity. The 1,1′-dimethyl-DIM derivative 71 (Ki 3.39 µM) reduced binding affinity, confirming a role for NH in interacting with the CB2 receptor. Substitution of the 2-position as in the 2,2′-dimethyl derivative 72 (Ki > 5 µM), or introduction of a nitrogen atom in the 7-position of the indole rings as in di-(7-azaindolyl)methane (73: Ki > 5 µM), led to a loss in CB2 receptor binding affinity. These results indicated that substituents at the 4,4′- or 6,6′-positions were well tolerated, and in some cases, especially those substituted in the 4,4′-positions, could lead to an improvement in binding affinity compared to lead structure 4. Concentration-inhibition curves for selected compounds are shown in Figure 2.

In the next set of experiments, we investigated the effects of substitution of the 3,3′-methylene bridge (C-10) of lead structure 4 (Table 2). A large variety of (aryl)alkyl substituents was introduced. Methyl substitution (100: Ki 5.74 µM) reduced binding affinity, while ethyl (101: Ki 0.804 µM) resulted in an affinity comparable to that of the lead compound. A further increase in the carbon chain length to propyl (102: Ki > 5 µM) and butyl (103: Ki > 5 µM) abolished affinity for the CB2 receptor. Introducing aryl substituents at the 3,3′-methylene bridge either reduced or abolished CB2 receptor affinity. Selected examples included p-tolyl (104: Ki 2.55 µM), m-tolyl (105: Ki 1.79 µM), o-tolyl (106: Ki 4.55 µM), p-anisyl (109: Ki 2.41 µM), m-anisyl (110: Ki 2.07 µM) and o-anisyl derivatives (111: Ki 2.72 µM), (4-phenoxy)phenyl (113: Ki > 5 µM), and a 4-hydroxyphenyl (114: Ki 7.13 µM)-substituted derivatives. The combination of substituents on the indole rings and on the methylene bridge of DIM did not improve binding affinity (see compounds 121–134, Table 2). Interestingly, a DIM derivative with a quaternary center (135: Ki 0.674 µM) showed an equipotent binding affinity to DIM, although the maximal displacement of radioligand binding was limited to 53% (see Table 2 and Figure 2b). However, its methoxyl derivative (136: Ki > 5 µM) lost the affinity for the CB2 receptor, indicating unfavorable interaction when a bulky substituent was present.
In summary, the substitution of the methylene bridge of DIM derivatives did not improve the compounds' binding affinity for the CB2 receptor, but a few derivatives retained moderate affinity.
As a next step, we investigated a series of unsymmetrically substituted DIMs for their binding to the CB2 receptor (see Table 3). Initially, only on one of both indole rings a substituent was introduced in the 4-position. The following rank order of potency was observed: 4-methyl-DIM (149: Ki 0.498 µM) > 4-F-DIM (150: Ki 0.758 µM) > 4-Br-DIM (151: Ki 0.944 µM). Thus, these compounds showed comparable binding affinity as the lead compound DIM. All of them were able to completely displace the radioligand in our experiments.
Next, mono-substitution at the 5-position of one of the indole rings was introduced. The following rank order of potency was observed: 5-Cl-DIM (154: Ki 1.21 µM) ≥ 5-methoxy-DIM (152: Ki 2.32 µM) ≥ 5-F-DIM (153: Ki3.78 µM). In general, mono-substitution at the 5-position appeared to be less advantageous than di-substitution on both indole rings in that position. We further investigated the behavior of unsymmetrical DIM derivatives with multiple substitutions, see compounds 157–170. As expected, substituents in the 4-position were found to yield the best unsymmetrical DIM derivatives. The following rank order of potency was observed: 4-Cl,4′-Br-DIM (157: Ki 0.237 µM) > 4-Cl,4′-CH3-DIM (158: Ki 0.536 µM). Both derivatives were able to nearly completely displace the radioligand. Compounds that have a 5-methoxy or 5-benzyloxy substituent combined with other substituents showed only moderate affinities (see compounds 159–168). 5-OCH3,4′-F-DIM (159: Ki 1.17 µM), and 5-OCH3,5′-CH3-DIM (164: Ki 1.20 µM) were among the most potent ligands in this series showing complete displacement of the radioligand. Introducing a polar hydroxyl group at position 5 of the indole ring (165: Ki 12.0 µM) reduced binding affinity. The N-methylated compound lost binding affinity (169: Ki > 5 µM), but introducing a methyl group at position 4′ of 170 restored affinity (170: Ki 0.716 µM).
We extended the SARs of DIM derivatives by preparing unsaturated (175–177) and saturated 2-oxyindole derivatives (178–180; Table 4). Among them, compound 176 showed moderate binding affinity, but the other compounds lost affinity.
The SARs of DIM derivatives at the CB2 receptor is summarized in Figure 3. DIM derivatives represent a new class of CB2 receptor ligands with Ki values reaching the submicromolar concentration range. In general, symmetrically substituted DIMs without substitutions on the methylene bridge showed similar or higher affinities as compared to the lead compound DIM. In particular, the presence of halogen at position 4 of both indole rings improved the binding affinity, while aryl substituents at the methylene bridge reduced it. At least one unsubstituted indole-NH appears to be required for CB2 receptor binding.

Some of the DIM derivatives, even very potent ones, showed incomplete inhibition of [3H]CP55,940 binding, see, for example, 51, 56, 136 (Tables 1 and 2, Figure 2). This may indicate that the DIM derivatives do not bind to the same binding site as the radioligand, that is the orthosteric binding site, but act as allosteric agonists. Another explanation could be that they do bind—at least in part—to the orthosteric binding site, but to a conformation that differs from the conformation to which classical CBs are binding or which they are stabilizing.
2.2.2 Functional assays at CB2 receptors
Next, we studied the functional activity of selected ligands. First, we performed cAMP accumulation assays as well as β-arrestin recruitment assays for DIM at the CB2 receptor to confirm previous results.[34] DIM behaved as a partial agonist in both cAMP and β-arrestin assays with EC50 values of 0.334 µM (Emax 65% compared to 1 µM full agonist of CP55,940) and 0.562 µM (Emax 63% compared to 0.1 µM full agonist of CP55,940) (see Figures 4 and 5, Table 5). Thus, DIM behaved similarly as the partial agonist THC, which displayed Emax values of 59% (cAMP) and 32% (β-arrestin assay), respectively.


| Human CB2 receptor | |||||
|---|---|---|---|---|---|
| Radioligand binding assay | cAMP assay | β-Arrestin recruitment assay | |||
| Ki ± SEM (µM) | EC50 ± SEM (µM) | EC50 ± SEM (µM) | |||
| (or percent inhibition of [3H]CP55,940 at 5 µM) | (or percent receptor activation at 10 µM) | (or percent receptor activation at 10 µM) | |||
| Compound | [Maximal inhibition (%)] | [efficacy]a | [efficacy]b | ΔΔlog (Emax/EC50)c | |
| 1 | THC | 0.00595 ± 0.00027 | 0.00527 ± 0.00019 | 0.00142 ± 0.00003 | 0.8 |
| [100%] | [59%] | [32%] | |||
| 2 | CP55,940 | 0.000293 ± 0.00008 | 0.00320 ± 0.00068 | 0.00262 ± 0.00003 | 0 |
| [100%] | [100%] | [100%] | |||
| 4 DIM | 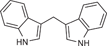 |
0.690 ± 0.159 | 0.334 ± 0.174 | 0.562 ± 0.195 | 0.3 |
| [98%] | [65%] | [63%] | |||
| 42 | 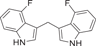 |
0.279 ± 0.056 | 0.0551 ± 0.0189 | 0.290 ± 0.148 | 0.9 |
| [99%] | [80%] | [70%] | |||
| 44 |  |
0.374 ± 0.074 | 0.509 ± 0.100 | 0.0450 ± 0.0189 | −0.8 |
| [100%] | [85%] | [61%] | |||
| 46 | 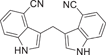 |
0.339 ± 0.061 | 0.0144 ± 0.0023 | 0.0149 ± 0.0021 | 0.2 |
| [99%] | [95%] | [67%] | |||
| 149 | 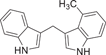 |
0.498 ± 0.176 | 0.0652 ± 0.0112 | 1.08 ± 0.37 | 1.3 |
| [98%] | [89%] | [93%] | |||
| 157 | 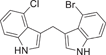 |
0.237 ± 0.006 | 0.237 ± 0.081 | 0.0385 ± 0.0125 | −0.6 |
| [98%] | [87%] | [69%] | |||
| 158 | 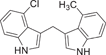 |
0.536 ± 0.058 | 0.281 ± 0.109 | 0.120 ± 0.045 | 0.2 |
| [96%] | [110%] | [51%] | |||
| 170 | 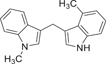 |
0.716 ± 0.001 | 0.228 ± 0.030 | 0.0803 ± 0.0340 | −0.3 |
| [99%] | [89%] | [70%] | |||
- a Efficacy relative to the maximal effect of the standard agonist CP55,940 at 1 µM set at 100%.
- b Efficacy relative to the maximal effect of the standard agonist CP55,940 at 0.1 µM set at 100%.
- c Bias factor was calculated as described in experimental section. Negative numbers indicate β-arrestin-biased compounds; positive numbers indicate G protein-biased agonists. The bias factor is logarithmic: 1 corresponds to a 10-fold bias, and two corresponds to a 100-fold bias.
Results for compounds selected for cAMP accumulation studies are shown in Figure 4. Like DIM, all investigated DIM derivatives behaved as partial agonists at the CB2 receptor, but their efficacy differed, some of the new compounds being more efficacious than DIM and significantly more efficacious than THC. Concentration-response curves were determined for compounds that showed more than 50% receptor activation at a concentration of 5 µM (see Figure 5). The results are collected in Table 5.
In the cAMP assay, the symmetrical substitution of DIM with 4,4′-difluoro-DIM (42: EC50 0.0551 µM) led to an increase in agonistic activity. The larger substituents in 4,4′-dibromo-DIM (44: EC50 0.509 µM) resulted in reduced activity at the CB2 receptor. Surprisingly, the 4,4′-dicyano-DIM derivative 46 (EC50 0.0144 µM) showed low nanomolar potency; in fact, compound 46 is the most potent agonist identified in the present series showing full intrinsic activity at the CB2 receptor. The mono-substituted 4-methyl DIM (149) displayed slightly lower activity at the CB2 receptor with an EC50 of 0.0652 µM. Reduced potency was found with unsymmetrically substituted DIM derivatives indicating that unsymmetrical substitution was not favorable for agonistic activity at the CB2 receptor. Concentration-response curves for selected compounds are depicted in Figure 5.
We further evaluated these selected compounds in β-arrestin recruitment assays using an enzyme complementation assay. The standard agonist CP55,940 was used as a reference agonist. Like in the cAMP assays, our lead compound DIM (4) showed partial agonistic activity in the β-arrestin recruitment assays as well (EC50 0.562 µM, 63% maximal activation). Thus, DIM showed nearly identical EC50 values in both the cAMP and the β-arrestin assays (Table 5). 4,4′-Difluoro-DIM (42: EC50 0.290 µM) exhibited an increase in agonistic activity compared to lead compound 4 (Figure 5). Surprisingly, introducing larger substituents like 4,4′-dibromo (44) 4,4′-dicyano (46), or 4-chloro,4′-bromo (157) increased the potency of the DIM derivatives in the β-arrestin assays dramatically yielding EC50 values of 0.0450, 0.0149, and 0.0385 µM, respectively.
We calculated bias factors for the most potent agonists, comparing their effects in Gi-dependent cAMP assays with those determined in β-arrestin assays, as previously described.[40, 41] CP55,940 was used as a reference compound (Table 5). A bias factor of 0 means no bias between two pathways, whereas a factor of 1 corresponds to a 10-fold preference, and a bias factor of two corresponds to a 100-fold preference for the G protein-dependent pathway. The partial agonist THC showed a slight preference for cAMP over β-arrestin signaling in our assay system (bias factor 0.8- or 6-fold preference for cAMP compared to β-arrestin signaling). This finding is well in agreement with published data.[42, 43] DIM (4) displayed a two-fold preference (bias factor 0.3) for cAMP over β-arrestin signaling. Compound 42 (4,4′-difluoro-DIM) showed a seven-fold preference (bias factor 0.9) for inhibition of cAMP accumulation, while 4,4′-dibromo-DIM (44) had the opposite preference being biased toward the β-arrestin over the cAMP pathway (bias factor −0.8). 4,4′-Dicyano-DIM (46) was characterized as a virtually unbiased agonist (bias factor: 0.2). In fact, 46 was found to be the most potent non-biased CB2 receptor agonist of the present series. The asymmetrical DIM-derivative 149 displayed the strongest bias towards cAMP over β-arrestin signaling, preferably activating Gi-dependent cAMP over β-arrestin signaling (bias factor: 1.3- or 19-fold preference). In contrast, unsymmetrically disubstituted DIM derivatives showed a preference for β-arrestin signaling over Gi-dependent cAMP signaling. This may result in functional antagonistic activity depending on the employed concentration since receptor internalization can be expected as a result of β-arrestin recruitment.
Next, we calculated the correlation of the results obtained in different experiments. The correlation between the potency of the DIM derivatives in cAMP and β-arrestin assays (Figure 6a) showed a low correlation. Some agonists are somewhat Gi protein-biased, which would result in longer-lasting activity because receptor internalization is less pronounced, while others are β-arrestin biased, which will lead to fast receptor internalization. Additionally, a comparison of functional and radioligand binding data revealed only a very low correlation (Figure 6b). This may be explained by the fact that DIM derivatives bind to a different binding site than the orthosteric agonist radioligand. Thus, DIM derivatives appear to act as allosteric CB2 receptor agonists.

2.2.3 Selectivity studies
Radioligand binding studies at the CB1 receptor subtype were performed and compared with results at the CB2 receptor (see Tables 1–4). The results showed that many of the compounds were able to bind to the CB1 receptor as well. Among them, 6,6′-di-Cl-DIM (62, CB1: Ki 0.820 µM; CB2: Ki 0.911 µM) was found to display comparable binding affinity at both CB1 and CB2 receptors. However, only a few compounds displayed CB1 receptor selectivity. Selected examples include 58 (Ki 2.95 µM), 119 (Ki 0.983 µM), 122 (Ki 0.541 µM), 123 (Ki > 0.414 µM), and 136 (Ki > 2.84 µM). Among these, compound 122 might be further developed in the future to obtain potent, selective CB1 receptor ligands. Importantly, the most potent CB2 receptor ligands, including 4, 42, 44, 46, 149, 157, 158, and 170, were selective for the CB2 versus the CB1 receptor (Supporting Information: Table S2, Figure 7). In contrast to THC, which is nonselective (see Figure 7), potent, CB2-selective agonists were discovered among the developed DIM derivatives. DIM (4) itself showed 8-fold selectivity for the CB2 versus the CB1 receptor. Compounds 42 (4,4′-di-F-DIM) and 46 (4,4′-di-CN-DIM) displayed the highest selectivity, being 36- and 30-fold selective for the CB2 receptor, respectively. Unsymmetrically substituted DIMs such as 149, 157, 158, and 170 showed a slightly lower CB2-selectivity index.

It is important to note that compound 70 (PSB-16671), a potent GPR84 allosteric agonist (EC50 0.043 µM) identified by our group, was shown to bind to both CB1 (Ki 1.10 µM) and CB2 receptors (Ki 2.64 µM) with comparable affinities. Mancini et al. recently reported that the ability of PSB-16671 to activate G proteins in mouse bone marrow-derived neutrophils was due to “off-target effects” and not mediated by GPR84.[44] This result was further supported by the inability of a GPR84 antagonist to block the effects of PSB-16671 on mouse GPR84 in both transfected cells and in the RAW264.7 cell line. Based on our results that PSB-16671 is able to interact with both CB receptor subtypes, we suggest that the observed effects of PSB-16671 could be due to its interaction with CB receptors natively expressed in the employed cell lines.
In native, nontransfected CHO cells DIM and its derivatives did not show any inhibition of cAMP accumulation. This clearly shows that the observed effects of DIM and its derivatives observed in the present study are due to the activation of the recombinantly expressed CB receptors in CHO cells. SARs at CB2 receptors and GPR84 are quite different, and both, CB2-selective or GPR84-selective agonists could be developed. However, one has to carefully choose concentrations—if high concentrations are employed, selectivity may not be given anymore.
We further investigated several DIM derivatives in functional assays at CB1 receptors employing cAMP accumulation as well as β-arrestin recruitment assays (Table 6, Figure 8, Supporting Information: Table S3). Interestingly, none of the compounds was active in cAMP assays at concentrations up to 10 µM, neither as agonist nor as antagonist versus the standard agonist CP55,940 (see Supporting Information: Table S3). However, they showed antagonistic activity in β-arrestin assays at micromolar concentrations (Table 6, Figure 8). This indicates once more that DIM derivatives can act as allosteric modulators, not only of CB2 but also of CB1 receptors. Depending on their substitution pattern, they can, in fact, act as biased CB1 receptor antagonists.
| Human CB1 receptor | |||
|---|---|---|---|
| Radioligand binding assay | cAMP assay (agonistic activity) | β-Arrestin recruitment assay (antagonistic activity) | |
| Ki ± SEM (µM) | EC50 ± SEM (µM) | IC50 ± SEM (µM) | |
| (or percent inhibition of [3H]CP55,940 at 5 µM) | (or percent receptor activation at 10 µM) | (or percent receptor inhibition at 10 µM) | |
| Compound | [Maximal inhibition (%)] | [efficacy]a | [Maximal inhibition (%)]b |
| THC | 0.00387 ± 0.00091 | 0.00326 ± 0.00017 | n.dc |
| [51%] | |||
| CP55,940 | 0.00192 ± 0.00140 | 0.00336 ± 0.00057 | n.d |
| [100%] | [100%] | ||
| 43 | 0.753 ± 0.048 | >10 (2%) | ≥10 (43%) |
| [61%] | |||
| 62 | 0.820 ± 0.385 | >10 (11%) | >10 (35%) |
| [59%] | |||
| 109 | 0.774 ± 0.169 | >10 (1%) | 6.09 ± 0.50 |
| [71%] | [91%] | ||
| 113 | 0.402 ± 0.306 | >10 (0%) | 4.43 ± 0.61 |
| [80%] | [94%] | ||
| 122 | 0.541 ± 0.173 | >10 (0%) | 3.06 ± 0.29 |
| [88%] | [91%] | ||
| 123 | 0.414 ± 0.260 | >10 (−10%) | 6.43 ± 0.45 |
| [68%] | [95%] | ||
- a Efficacy relative to the maximal effect of the standard agonist CP55,940 at 1 µM set at 100%.
- b Inhibition compared to the EC80 of CP55,940 (0.001 µM) at the human CB1 receptor, set at 100%.
- c n.d, not determined.

3 CONCLUSIONS
In conclusion, a series of 99 symmetrical and unsymmetrical DIM derivatives and analogs were synthesized and evaluated with the aim to optimize their CB2 receptor affinity, selectivity, and efficacy, of which 44–46, 50, 105, 106, 108, 110, 111, 113, 119, 124, 125, 135, 136, and 165 are new compounds not previously reported in the literature. Compounds 42, 44, 46, 149, 157, 158, and 170 displayed high CB2 receptor binding affinity and selectivity versus the CB1 receptor. When they were investigated in functional assays, namely cAMP accumulation, and β-arrestin recruitment assays, the compounds behaved as CB2 receptor agonists. However, only low correlations between affinity determined in binding assays and potency measured in functional assays were observed strongly hinting at allosteric interactions. Some of the compounds showed biased signaling either for G protein-dependent cAMP production or for β-arrestin recruitment. Di-(4-cyano-1H-indol-3-yl)methane (46, PSB-19837, EC50 [cAMP]: 0.0144 µM, EC50 [β-arrestin]: 0.0149 µM) was the most potent unbiased CB2 receptor agonist. On the other hand, di-(4-bromo-1H-indol-3-yl)methane (44, PSB-19571, EC50 (cAMP): 0.509 µM, EC50 (β-arrestin): 0.0450 µM) is biased toward β-arrestin signaling, while 3-((1H-indol-3-yl)methyl)-4-methyl-1H-indole (149, PSB-18691, EC50 [cAMP]: 0.0652 µM, EC50 [β-arrestin]: 1.08 µM) is a Gi-protein biased CB2 receptor agonist. These tool compounds possessing different pharmacological profiles will be useful for studying CB2 receptors. Few DIM derivatives of the present series showed moderate affinity and potency at the CB1 receptor, inhibiting CB1-mediated β-arrestin recruitment (109, 113, 122, and 123). Our results clearly point to an allosteric binding site for DIM derivatives at CB1 and CB2 receptors, and the determined radioligand binding data actually underestimate the compounds' potency in most cases. These DIM derivatives can serve as valuable tool compounds to elucidate the role of different signaling pathways activated by CB2 receptors.
4 EXPERIMENTAL
4.1 Chemistry
4.1.1 General
All commercially available reagents were used as purchased (Acros, Alfa Aesar, Sigma-Aldrich, ABCR or TCI). Solvents were used without additional purification or drying except for dichloromethane, which was distilled over calcium hydride. Thin layer chromatography (TLC) using aluminum sheets with silica gel 60 F254 was employed to monitor the reactions (Merck). Column chromatography was performed with silica gel 0.060–0.200 mm, pore diameter ca. 6 nm. For microwave reactions, a CEM-Focused Microwave Synthesis type Discover apparatus was used. All synthesized compounds were finally dried in a vacuum at 8–12 Pa (0.08–0.12 mbar) using a sliding vane rotary vacuum pump (Vacuubrand GmbH). 1H-, 13C NMR, and 13Capt NMR data were collected on a Bruker Avance 500 MHz NMR spectrometer at 500 MHz (1H), or 126 MHz (13C), respectively. If indicated, NMR data were collected on a Bruker Ascend 600 MHz NMR spectrometer at 600 MHz (1H), or 151 MHz (13C), respectively. DMSO-d6 was employed as a solvent at 303 K unless otherwise noted. Chemical shifts are reported in parts per million (ppm) relative to the deuterated solvent; that is, DMSO, δ 1H: 2.49 ppm; 13C: 39.7 ppm. Coupling constants J are given in Hertz and spin multiplicities are given as s (singlet), d (doublet), t (triplet), q (quartet), sext. (sextet), m (multiplet), br (broad). Melting points were determined on a Büchi 530 melting point apparatus and are uncorrected. The purities of isolated products were determined by ESI-mass spectra obtained on an LCMS instrument (Applied Biosystems API 2000 LCMS/MS, HPLC Agilent 1100) using the following procedure: the compounds were dissolved at a concentration of 1.0 mg/ml in acetonitrile containing 2 mM ammonium acetate. Then, 10 μl of the sample were injected into an HPLC column (Macherey-Nagel Nucleodur® 3 µl C18, 50 × 2.00 mm). Elution was performed with a gradient of water/acetonitrile (containing 2 mM ammonium acetate) from 90:10 to 0:100 for 20 min at a flow rate of 300 μl/min, starting the gradient after 10 min. UV absorption was detected from 200 to 950 nm using a diode array detector. Purity of all compounds was determined at 254 nm. The purity of the compounds was generally ≥95%. Compounds 44–46, 50, 105, 106, 108, 110, 111, 113, 119, 124, 125, 135, 136, and 165 are new, and not previously reported in the literature.
The InChI codes of the investigated compounds, together with some biological activity data, are provided as Supporting Information.
4.1.2 General procedure for the synthesis of diindolylmethane derivatives
General procedure for the preparation of 4, 40–73[33]
The appropriate indole (5–41, 10 mmol) and formaldehyde (38%) (5 mmol) in water (5 ml) were microwave-irradiated for the required period of time at 100°C. The mixture was diluted with water (50 ml) and extracted with ethyl acetate (2 × 50 ml) after the reaction was completed. The combined organic layers were rinsed with brine, dried over MgSO4, filtered, and evaporated to dryness under reduced pressure. The crude product was purified by silica gel column chromatography. Compounds 4, 40–43, 47–49, 51–62, 63, 64–73 were previously reported.[33]
Di[4-bromo-indol-3-yl]methane (44)
The compound was synthesized by reaction of 4-bromoindole (10, 10 mmol) with formaldehyde (38%) (5 mmol) in water (5 ml). The product was isolated as a brown solid (82% yield, 166 mg). 1H NMR (600 MHz, DMSO-d6) δ 11.31 (S, 2H, NH), 7.37 (dd, J = 8.1, 0.9 Hz, 2H, Ar), 7.13 (dd, J = 7.6, 0.8 Hz, 2H, Ar), 6.95 (t, J = 7.8 Hz, 2H, Ar), 6.85 (dd, J = 2.0, 0.9 Hz, 2H, Ar), 4.79–4.41 (m, 2H, CH2). 13C NMR (151 MHz, DMSO) δ 138.12 (C8), 125.71 (C7), 124.84 (C5), 122.58 (C6), 122.08 (C2), 115.55 (C4), 113.37 (C7), 111.38 (C3), 23.56 (CH2). LC-MS (m/z): positive mode 405 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 96.0%.
Di[4-nitro-indol-3-yl]methane (45)
The compound was synthesized by reaction of 4-nitroindole (11, 10 mmol) with formaldehyde (38%) (5 mmol) in water (5 ml). The product was isolated as a brown solid (69% yield, 116 mg). 1H NMR (500 MHz, DMSO-d6) δ 8.09 (dd, J = 8.1, 6.3 Hz, 2H, Ar), 7.81 (d, J = 3.2 Hz, 2H, Ar), 7.37 (t, J = 8.0 Hz, 1H, Ar), 7.04 (d, J = 3.2 Hz, 1H, Ar), 6.65 (t, J = 7.3 Hz, 2H, Ar), 4.21 (d, J = 7.4 Hz, 2H, CH2).13Capt NMR (126 MHz, DMSO-d6) δ 139.48 (C4), 137.73 (C8), 122.41 (C2), 120.69 (C6), 118.37 (C7), 117.52 (C5 and C7), 101.11 (C3), 29.33 (CH2). LC-MS (m/z): positive mode 337 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 97.0%.
Di[4-cyano-indol-3-yl]methane (46)
The compound was synthesized by reaction of 4-cyanoindole (12, 10 mmol) with formaldehyde (38%) (5 mmol) in water (5 ml). The product was isolated as a yellow solid (69% yield, 102 mg). 1H NMR (600 MHz, DMSO-d6) δ 11.62 (s, 2H, NH), 7.72 (dd, J = 8.2, 0.8 Hz, 2H, Ar), 7.47 (dd, J = 7.3, 0.8 Hz, 2H, Ar), 7.22 (dd, J = 8.2, 7.4 Hz, 2H, Ar), 7.14 (d, J = 2.1 Hz, 1H, Ar), 4.73 (s, 2H, CH2). 13C NMR (151 MHz, DMSO-d6) δ 136.94 (C9), 127.22 (C7), 125.86 (C5), 121.03 (C2), 119.51 (C6), 117.19 (CN), 114.02 (C7), 100.69 (C3), 20.60. LC-MS (m/z): positive mode 297 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 96.0%.
Di[5-trifluoromethylindol-3-yl]methane (50)
The compound was synthesized by reaction of 5-trifluoromethylindole (16, 10 mmol) with formaldehyde (38%) (5 mmol) in water (5 ml). The product was isolated as a yellow solid (78% yield, 149 mg). 1H NMR (600 MHz, DMSO-d6) δ 10.82 (s, 2H, NH), 7.32–7.26 (m, 3H, Ar), 7.26 (d, J = 2.3 Hz, 2H, Ar), 7.21 (dd, J = 10.1, 2.5 Hz, 2H, Ar), 6.85 (dd, J = 9.2, 2.6 Hz, 2H, Ar), 4.05 (s, 2H, CH2). 13C NMR (151 MHz, DMSO-d6) δ 133.20 (C9), 127.46 (C8), 127.40 (CF3), 125.12 (C5), 118.37 (C2), 114.34 (C4), 112.38 (C6), 112.32 (C7), 108.88 (C3), 20.86 (CH2). LC-MS (m/z): positive mode 383 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 96.0%.
General procedure for the synthesis of 100–136[33, 44]
Concentrated sulfuric acid (1 equiv.) was added to a stirred mixture of the suitable indole (5, 7, 8, 13–15, 27, 30, or 32; 3.1–6.7 mmol) and the appropriate aldehyde or ketone (74–99; 1.5–3.3 mmol, 0.5 equiv.) diluted in water (5 ml). The aqueous suspension was dissolved in ethyl acetate and rinsed with brine once the reaction was completed. The organic layer was dried over MgSO4, filtered, and evaporated to dryness under reduced pressure. Silica gel column chromatography was used to purify the crude product. Compounds 100–104,[33] 107,[45] 109,[35] 112, [33] 114-117, [33] 118,[45] 120,[33] 121–122,[45] 123,[33] 126–134[45] were previously reported.
3,3'-(m-Tolylmethylene)di(indole) (105)
The compound was synthesized by reaction of indole (5, 3.1 mmol) with 3-methylbenzaldehyde (79, 1.5 mmol) in water (5 ml). The product was isolated as a brown solid (76% yield, 397 mg). 1H NMR (500 MHz, DMSO-d6) δ 10.75 (s, 2H), 7.36–7.31 (m, 2H, Ar), 7.27 (d, J = 7.9 Hz, 2H, Ar), 7.18 (s, 1H, Ar), 7.13 (dd, J = 4.0, 1.1 Hz, 2H), 7.02 (dd, J = 8.1, 7.1 Hz, 2H, Ar), 7.00–6.93 (m, 1H, Ar), 6.85 (dd, J = 8.0, 7.1 Hz, 2H, Ar), 6.80 (dd, J = 8.5, 6.7 Hz, 2H, Ar), 5.77 (s, 1H, CH-), 2.23 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ 145.06 (C-3"), 137.01 (C-1"), 136.68 (C9), 129.04 (C8), 128.00 (C-2"), 126.77 (C-5"), 126.58 (C-4"), 125.49 (C-6"), 123.63 (C5), 120.94 (C6 and C4), 119.18 (C2), 111.53 (C3), 40.26 (C10), 21.16 (CH3). LC-MS (m/z): positive mode 337 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 97.0%.
3,3′-(o-Tolylmethylene)di(indole) (106)
The compound was synthesized by reaction of indole (5, 3.1 mmol) with 2-methylbenzaldehyde (80, 1.5 mmol) in water (5 ml). The product was isolated as a brown solid (79% yield, 412 mg). 1H NMR (500 MHz, DMSO-d6) δ 10.75 (s, 2H, NH), 7.34 (dd, J = 8.1, 0.8 Hz, 2H, Ar), 7.21 (d, J = 7.9 Hz, 2H, Ar), 7.16 (dd, J = 7.3, 0.7 Hz, 1H, Ar), 7.07 (dd, J = 7.9, 6.5 Hz, 2H, Ar), 7.02 (dd, J = 8.1, 5.2 Hz, 3H, Ar), 6.84 (dd, J = 8.0, 7.1 Hz, 2H, Ar), 6.66 (d, J = 1.7 Hz, 2H, Ar), 5.94 (s, 1H, CH), 2.33 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ 143.91 06 (C-2"), 136.77 06 (C-1"), 135.50 06 (C-9), 130.15 06 (C-8), 127.96 (C-5"), 126.84 (C-4"), 125.85 (C-6"), 125.62 (C-5), 124.03 (C-2), 120.97 (C-5), 119.02 (C-6), 118.29 (C-4), 111.58 (C-3), 40.29 (C10), 19.26 (CH3). LC-MS (m/z): positive mode 337 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 96.0%.
3,3′-[(4-Isopropylphenyl)methylene]di(indole) (108)
The compound was synthesized by reaction of indole (5, 3.1 mmol) with 4-isopropylbenzaldehyde (82, 1.5 mmol) in water (5 ml). The product was obtained as a brown solid (68% yield, 385 mg). 1H NMR (500 MHz, CDCl3) δ 7.87 (s, 2H, NH), 7.39 (d, J = 8.0 Hz, 2H, Ar), 7.33 (d, J = 8.2 Hz, 2H, Ar), 7.25 (d, J = 7.6 Hz, 2H, Ar), 7.15 (d, J = 7.3 Hz, 2H, Ar), 7.14–7.08 (m, 2H, Ar), 6.99 (t, J = 7.5 Hz, 2H, Ar), 6.65 (s, 2H, Ar), 5.84 (s, 1H, CH-), 2.97–2.72 (m, 1H, CH), 1.22 (d, J = 6.9 Hz, 6H, 2CH3). 13C NMR (126 MHz, CDCl3) δ 146.44 (C-4"), 141.21 (C-9), 136.67 (C-8), 128.49 (C-2"), 127.13 (C-6"), 126.18 (C-3" and C-5"), 123.52 (C-2), 121.83 (C-6), 119.98 (C-5), 119.12 (C-4), 110.96 (C3, and C7), 39.74 (C10), 33.71 (CH), 20.63 (2 x CH3). LC-MS (m/z): positive mode 365 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 98.0%.
3,3′-[(3-Methoxyphenyl)methylene]di(indole) (110)
The compound was synthesized by reaction of indole (5, 3.1 mmol) with 3-methoxybenzaldehyde (84, 1.5 mmol) in water (5 ml). The product was obtained as a brown solid (82% yield, 449 mg). 1H NMR (500 MHz, DMSO) δ 10.77 (s, 2H, NH), 7.33 (d, J = 8.1 Hz, 2H, Ar), 7.28 (d, J = 7.9 Hz, 2H, Ar), 7.17 (t, J = 7.9 Hz, 1H, Ar), 7.02 (t, J = 7.5 Hz, 2H, Ar), 6.91 (dd, J = 12.5, 5.0 Hz, 2H, Ar), 6.88–6.80 (m, 4H, Ar), 6.74 (d, J = 2.6 Hz, 1H, Ar), 5.81 (d, J = 19.6 Hz, 1H, CH-), 3.66 (s, 3H, OCH3). 13C NMR (126 MHz, DMSO) δ 159.23 (C-3"), 146.77 (C-1"), 136.69 (C-5"), 129.08 (C-9), 126.77 (C8), 123.63 (C6), 120.96 (C2), 119.20 (C-6"), 118.43 (C4), 118.04 (C5), 114.63 (C-2"), 111.54 (C-4"), 110.79 (C3), 54.97 (CH, CH3). LC-MS (m/z): positive mode 353 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 96.0%.
3,3′-[(2-Methoxyphenyl)methylene]di(indole) (111)
The compound was synthesized by reaction of indole (5, 3.1 mmol) with 2-methoxybenzaldehyde (85, 1.5 mmol) in water (5 ml). The product was obtained as a brown solid (79% yield, 432 mg). 1H NMR (500 MHz, DMSO) δ 10.70 (s, 2H, NH), 7.32 (d, J = 8.1 Hz, 2H, Ar), 7.20 (d, J = 7.9 Hz, 2H, Ar), 7.18–7.05 (m, 2H, Ar), 7.04–6.95 (m, 3H, Ar), 6.83 (dd, J = 7.6, 0.9 Hz, 2H, Ar), 6.80 (dd, J = 7.4, 1.0 Hz, 1H, Ar), 6.71 (d, J = 1.7 Hz, 2H, Ar), 6.20 (s, 1H, CH-), 3.79 (s, 3H, OCH3). 13C NMR (126 MHz, DMSO) δ 156.42 (C-2"), 136.70 (C-9), 132.81 (C-6"), 129.24 (C-1"), 127.10 (C-4"), 126.88 (C-8), 123.67 (C-2), 120.88 (C-5"), 120.15 (C-4), 119.00 (C-6), 117.93 (C-5), 111.51 (C-3" and C-3), 55.72 (OCH3). LC-MS (m/z): positive mode 353 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 96.0%.
3,3′-[(4-Phenoxyphenyl)methylene]di-(indole) (113)
The compound was synthesized by reaction of indole (5, 3.1 mmol) with 4-phenoxybenzaldehyde (87, 1.5 mmol) in water (5 ml). The product was obtained as a yellow solid (71% yield, 387 mg). 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 2.4 Hz, 2H, Ar), 7.43–7.37 (m, 2H, Ar), 7.34 (dd, J = 8.2, 0.9 Hz, 2H, Ar), 7.32–7.26 (m, 4H, Ar), 7.17 (dd, J = 8.2, 7.0 Hz, 2H, Ar), 7.11–7.03 (m, 1H, Ar), 7.03–6.95 (m, 4H, Ar), 6.95–6.89 (m, 2H, Ar), 6.65 (dd, J = 2.4, 1.0 Hz, 2H, Ar), 5.86 (s, 1H, CH). 13C NMR (126 MHz, CDCl3) δ 157.48 (C-1’’′), 155.30 (C-4"), 139.01 (C-9), 136.69 (C-1"), 129.90 (C-2"), 129.62 (C-6"), 127.00 (C-3’’′ and 5’’′), 123.52 (C2), 122.93 (C-3"), 121.95 (C-5"), 119.91 (C-6), 119.75 (C-2’’′ and 6’’′), 118.73 (C7), 118.61 (C4), 111.05 (C3), 60.40 (C10). LC-MS (m/z): positive mode 415 [M+H]1+; Purity by HPLC-U (254 nm)-ESI-MS: 96.0%.
3,3′-[(7-Methoxynaphth-1-yl)methylene]di(indole) (119)
The compound was synthesized by reaction of indole (5, 3.1 mmol) with 7-methoxy-1-naphthaldehyde (93, 1.5 mmol) in water (5 ml). The product was obtained as a brown solid (89% yield, 556 mg).1H NMR (500 MHz, CDCl3) δ 7.94–7.86 (s, 2H, NH), 7.70–7.61 (m, 2H, Ar), 7.59 (d, J = 8.9 Hz, 1H, Ar), 7.46 (dd, J = 8.4, 1.8 Hz, 1H, Ar), 7.42–7.37 (m, 2H, Ar), 7.34 (dd, J = 8.2, 0.8 Hz, 2H, Ar), 7.23 (s, 1H, Ar), 7.15 (dd, J = 8.2, 7.0 Hz, 2H, Ar), 7.10 (d, J = 2.6 Hz, 1H, Ar), 7.07 (dd, J = 8.9, 2.5 Hz, 1H, Ar), 6.97 (dd, J = 8.0, 7.1 Hz, 2H, Ar), 6.65 (dd, J = 2.2, 1.0 Hz, 2H, Ar), 6.01 (s, 1H, CH-), 3.89 (s, 3H, OCH3). 13C NMR (126 MHz, CDCl3) δ 157.29 (C-OCH3), 139.31 (C9), 136.70 (naphthyl), 133.32 (naphthyl), 129.37 (naphthyl), 129.03 (naphthyl), 128.22 (C8), 127.11 (naphthyl), 126.60 (naphthyl), 126.59 (naphthyl), 123.72 (C2), 121.92 (C6), 119.98 (C5), 119.96 (C4), 119.74 (naphthyl), 110.99 (C7), 105.64 (C3), 55.29 (C10), 40.08 (OCH3). LC-MS (m/z): positive mode 403 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 96.0%.
3,3′-[(4-Fluorophenyl)methylene]di(5-methoxyindole) (124)
The compound was synthesized by reaction of 5-methoxyindole (14, 3.1 mmol) with 4-methoxybenzaldehyde (83, 1.5 mmol) in water (5 ml). The product was obtained as a brown solid (82% yield, 510 mg). 1H NMR (500 MHz, DMSO-d6) δ 10.66 (d, J = 2.4 Hz, 2H, NH), 7.28–7.12 (m, 2H, Ar), 6.63 (s, 1H, Ar), 6.52 (dd, J = 2.4, 0.8 Hz, 2H, Ar), 6.35 (dd, J = 6.3, 2.3 Hz, 2H, Ar), 5.89 (s, 1H, CH), 3.59 (s, 6H, 2 x OCH3). 13C NMR (126 MHz, DMSO-d6) δ 161.55 (C-4’’′, J = 257 Hz), 155.66 (C5), 138.11 (C-1"), 130.02 (C-2" and C-6"), 129.95 (C-9), 122.25 (C8), 121.75 (C2), 120.17 (C-3" and C-5"), 116.75 (C6), 114.28 (C7), 114.12 (C3), 104.89 (C4), 55.13 (2 x OCH3). LC-MS (m/z): positive mode 401 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 99.0%.
3,3′-[(4-Methoxyphenyl)methylene]di(5-methoxyindole) (125)
The compound was obtained by reaction of 5-methoxyindole (14, 3.1 mmol) with 4-fluorobenzaldehyde (90, 1.5 mmol) in water (5 ml). The product was isolated as a brown solid (78% yield, 485 mg). 1H NMR (500 MHz, DMSO-d6) δ 10.58 (d, J = 2.5 Hz, 2H, NH), 7.23 (dd, J = 10.9, 8.7 Hz, 4H, Ar), 6.86–6.79 (m, 2H, Ar), 6.77 (dd, J = 2.4, 0.8 Hz, 2H, Ar), 6.71 (d, J = 2.5 Hz, 2H, Ar), 6.68 (dd, J = 8.7, 2.5 Hz, 2H, CH-), 3.70 (s, 3H, OCH3), 3.59 (s, 6H, 2 x OCH3). 13C NMR (126 MHz, DMSO-d6) δ 157.42 (C-4’’′), 152.75 (C5), 137.12 (C-1"), 131.96 (C9), 129.32 (C8), 127.13 (C-2" and C-6"), 124.30 (C2), 118.17 (C-3" and C-5"), 113.48 (C5), 112.06 (C6), 110.58 (C3), 101.73 (C7), 55.42 (C10), 55.07 (3 x OCH3). LC-MS (m/z): positive mode 413 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 99.0%.
3,3′-[1-(p-Tolyl)ethane-1,1-diyl]di(indole) (135)
The compound was synthesized by reaction of indole (5, 3.1 mmol) with 1-(p-tolyl)ethan-1-one (98, 1.5 mmol) in water (5 ml). The product was obtained as a brown solid (74% yield, 402 mg). 1H NMR (500 MHz, DMSO-d6) δ 10.73 (d, J = 1.8 Hz, 2H, Ar), 7.32 (s, 2H, Ar), 7.28–7.13 (m, 2H, Ar), 7.07 (d, J = 8.1 Hz, 2H, Ar), 7.02 (d, J = 8.0 Hz, 2H, 2H), 6.99–6.90 (m, 2H, Ar), 6.74 (dd, J = 8.0, 7.0 Hz, 2H, Ar), 6.65–6.63 (m, 1H, Ar), 3.30 (s, 3H, CH3), 2.24 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ 145.16 (C-1"), 137.19 (C9), 134.67 (C-4’’′), 128.69 (C8), 127.87 (C-3’’′ and C5’’′), 126.32 (C-2’’′ and C6’’′), 123.44 (C2), 123.20 (C6), 121.40 (C5), 120.52 (C4), 117.81 (C7), 111.82 (C3), 42.94 (C10), 28.86 (CH3). LC-MS (m/z): positive mode 351 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 96.0%.
3,3′-[1-(4-Methoxyphenyl)ethane-1,1-diyl]di(indole) (136)
The compound was synthesized by reaction of indole (5, 3.1 mmol) with 1-(p-anisyl)ethan-1-one (99, 1.5 mmol) in water (5 ml). The product was obtained as a brown solid (79% yield, 450 mg). 1H NMR (500 MHz, DMSO-d6) δ 10.72 (s, 2H, NH), 7.32 (d, J = 8.1 Hz, 2H, Ar), 7.27–7.15 (m, 2H, Ar), 7.07 (s, 2H, Ar), 7.00–6.91 (m, 2H, Ar), 6.83–6.76 (m, 2H, Ar), 6.75 (dd, J = 8.0, 0.9 Hz, 2H, Ar), 6.71 (dd, J = 8.6, 1.7 Hz, 3H, Ar), 165.3.70 (s, 3H, OCH3), 2.19 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ 157.13 (C-4’’′), 140.57 (C9), 137.20 (C-1’’′), 126.22 (C-2’’′ and C6’’′), 123.42 (C8), 123.30 (C2), 121.16 (C6), 120.53 (C5), 118.00 (C4), 112.96 (C-3’’′ and C5’’'), 111.65 (C3), 55.01 (OCH3), 42.62 (C10), 29.33 (C10). LC-MS (m/z): positive mode 367 [M+H]1+; Purity by HPLC-UV(254 nm)-ESI-MS: 97.0%.
General Procedure for the synthesis of 149–170[36]
Procedure A
(3-Indolylmethyl)trimethylammonium iodides (137–140, 1.6 mmol) and the appropriate indole derivative (6–8, 13–15, 17, 21, 27, 30, 37, 38, 141, 142, or 143; 3.2 mmol) were dissolved in H2O (5 ml) in a 50 ml sealed tube. The reaction mixture was heated at 80°C upon stirring. After completion of the reaction, which was monitored by TLC, the mixture was allowed to cool to room temperature. The compound that precipitated on the tube wall was dissolved in ethyl acetate (10 ml), after the water had been decanted from the mixture. The resulting solution was dried over Na2SO4 and the product was purified by recrystallization or column chromatography.
Procedure B
In a 50 ml sealed tube, the appropriate indole derivative (6–8, 13–15, 17, 21, 27, 30, 37, 38, 141, 142, or 143; 1.14 mmol) and Cu(OAc)2∙H2O (0.57 mmol) were given to a solution of a 2-(1H-indol-3-yl)acetic acid derivative (144–148; 0.57 mmol) in ACN (5 ml). The reaction mixture was stirred for 2 h at 115°C. The mixture was cooled to room temperature when the reaction was completed as monitored by TLC. The reaction mixture was poured into water and extracted with 2 × 25 ml of ethyl acetate. The mixed organic layers were washed with a brine solution (25 ml), dried over Na2SO4, and evaporated to dryness under reduced pressure. Recrystallization or column chromatography was used to purify the crude product.
Compounds 149–151,[36] 152–153,[36, 37] 154,[36] 155–156,[33, 36] 157–158,[36] 159–164,[37] 165–168,[37] 169,[36] and 170,[33] have previously been reported.
3-[(6-Fluoroindol-3-yl)methyl]indole-5-ol (165)
The compound was synthesized according to procedure B by reaction of 6-fluorolindole (27, 0.57 mmol) with 2-(5-hydroxy-1H-indol-3-yl)acetic acid (147, 0.57 mmol) in ACN (5 ml). The product was obtained as a brown solid (78% yield,125 mg). 1H NMR (600 MHz, DMSO-d6) δ 12.25 (s, 1H, NH), 11.09 (s, 1H, NH), 10.73 (s, 1H, OH), 7.66 (d, J = 8.4 Hz, 2H, Ar), 7.50 (d, J = 7.8 Hz, 1H, Ar), 7.36 (d, J = 8.5 Hz, 1H, Ar), 7.31 (d, J = 8.1 Hz, 1H Ar), 7.24 (s, 1H, Ar), 7.09 (s, 1H, Ar), 7.02 (t, J = 7.4 Hz, 1H, Ar), 6.91 (t, J = 7.4 Hz, 1H, Ar), 4.16 (s, 2H, Ar). 13C NMR (151 MHz, DMSO-d6) δ 168.62 (C6, J = 287 Hz), 139.04 (C-5′), 136.56 (C-9′), 127.24 (C9), 126.86 (C8), 124.59 (C8′), 122.88 (C2), 122.28 (C-2′), 120.95 (C-4′), 118.74 (C3), 118.21 (C5), 115.75 (C6), 113.99 (C-3′), 111.47 (C-5′), 111.15 (C-7′), 20.95 (C10). LC-MS (m/z): positive mode 281[M+H]1+; purity by HPLC-UV(254 nm)-ESI-MS: 96.0%.
General procedure for the synthesis of 175–177
2-Oxindole (171–172, 37 mmol) in absolute ethanol was treated with the appropriate indolecarboxaldehyde (173–174; 41 mmol) and piperidine (16 mmol). After heating the mixture at 65°C for 2 h, the solvent was evaporated under reduced pressure. The residue was dissolved in 50 ml of water and extracted with ethyl acetate (2 × 30 ml). The combined organic layer was washed with brine, dried over MgSO4, and evaporated to dryness. The product was purified by silica gel column chromatography. Compounds 175–177 have previously been reported.[33]
General procedure for the synthesis of 178–180
NaBH4 (12 mmol) was added portion-wise over a period of 5–10 min at room temperature to a solution of the appropriate 2-oxindole derivative (175–177, 10 mmol) in ethanol (20 ml), and the reaction mixture was heated at 65°C for 2 h. Once the reaction was completed, the mixture was cooled to rt, and the ethanol was evaporated under reduced pressure. The residue was dissolved in ice water and extracted with 2 × 50 ml of ethyl acetate. The organic layers were combined, washed with brine, dried over MgSO4, filtered, and evaporated under reduced pressure. The resulting product was purified by column chromatography using a mixture of petroleum ether and ethyl acetate. Compounds 178–180 have been described previously.[33]
4.2 Pharmacological/biological assays
4.2.1 Radioligand binding assays at CB1 and CB2 receptors
Competition binding assays were performed using the nonselective CB receptor agonist radioligand [3H](−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol ([3H]CP55,940, final concentration 0.1 nM) as previously described.[38, 46, 47] Membrane preparations of CHO cells stably expressing either human CB1 or CB2 receptor were used (CB1: 30 μg of protein/well and CB2: 16 µg of protein/well) for all of the radioligand binding experiments. Stock solutions of the DIM derivatives were prepared in DMSO. The mixture containing 15 μl of the test compound in DMSO, 60 μl of [3H]CP55,940 solution in assay buffer (50 mM TRIS, 3 mM MgCl2, 0.1% bovine serum albumin [BSA], pH 7.4), 60 μl of membrane preparation and 465 μl of assay buffer was incubated for 2 h at room temperature (final DMSO conc. in the assay was 2.5%). Incubation was terminated by filtration through GF/C glass fiber filters (presoaked for 0.5 h in 0.3% aq. polyethyleneimine solution). Total binding was determined by adding DMSO without test compound, while nonspecific binding was determined in the presence of 10 μM of unlabeled CP55,940. The filter was then dried for 1.5 h at 50°C. Radioactivity on the filters was determined in a liquid scintillation counter (Top count NXT, Packard/Perkin-Elmer) after 10 h of preincubation with 50 µl of scintillation cocktail (Multiscint 25, Perkin-Elmer). Data were obtained in minimum of three independent experiments, performed in duplicates. For the calculation of Ki values, the Cheng-Prusoff equation and KD values of 2.4 nM ([3H]CP55,940 at CB1) and 0.7 nM ([3H]CP55,940 at CB2) were used.[48]
4.2.2 cAMP accumulation assays at human CB1 and CB2 receptor
The inhibition of adenylate cyclase activity was determined according to a described procedure in the literature using a competition binding assay for cAMP quantification in CHO cells stably expressing the CB1 or the CB2 receptor subtype, respectively.[21, 30] Briefly, cells were seeded into a 24-well plate (200,000 cells/well) and incubated for 24 h. On the day of the assay, the medium was exchanged for Hank's buffered saline solution (HBSS, Gibco) and the cells were further incubated for another 2 h. About 20 µl of 40 µM Ro-20-1724 (4-(3-butoxy-4-methoxybenzyl)-2-imodazolidinone) as phosphodiesterase inhibitor was added into each of the wells. After 10 min incubation, 15 µl of test compound (diluted in HBSS) was added to the mixture and the cells were further incubated for another 5 min. The adenylate cyclase activator, forskolin (final concentration 10 µM), was added to the mixture, and incubation was continued for a further 15 min. The final DMSO concentration was 1.9%. The reaction was stopped by lysis of the cells with hot lysis buffer (100°C; 4 mM EDTA, 0.01% Triton X-100). The cAMP quantification was performed by mixing 50 μl of cell suspension, 30 μl of [3H]cAMP (3 nM in Tris buffer), and 40 μl of cAMP-binding protein (50 µg per well in Tris buffer). The mixture was incubated for 1 h on ice. Bound and free radioligand were separated through GF/B glass fiber filters, and the radioactivity was measured after 9 h of preincubation with a scintillation cocktail (LumaSafeplus, Perkin-Elmer). Data were obtained from three independent experiments, performed in duplicates.
4.2.3 β-Arrestin assays at human CB1 and CB2 receptor
β-Arrestin recruitment assays based on galactosidase enzyme complementation assay (DiscoverX) were performed according to previously published.[38] Briefly, CHO β-arrestin2 cells stably transfected either human CB1-prolink1 or human CB2-prolink1 were seeded in the density of 30,000 cells/well (CB1), or 20,000 cells/well (CB2), and incubated for 24 h. On the day of the assay, about 10 µl of the test compound was added and the cells were further incubated for another 90 min. CP 55,940 (0.1 µM) was used as the standard agonist for maximal response. For antagonistic activity, the test compounds (5 µl) were added to the wells, and incubated for 60 min at 37°C, and then the agonist at its EC80 was added (5 µl) and the mixture was incubated for another 90 min at 37°C. The galactosidase activity was measured by lysis of the cells according to the manufacturing protocol. The luminescence was measured with an LBMitras 940 plate reader (Berthold, Bad Wildbad). A minimum of three independent experiments was performed, each in duplicate.
4.2.4 Operational model to determine the bias factor of the agonists
The pathway bias was calculated as described by Winpenny et al.[41, 49] and Pillaiyar et al.[40] The Emax (max activation in %) and the EC50 (in M) for each compound were used to obtain the transduction ratio (log (Emax/EC50). The transduction ratio within a pathway (Δlog(Emax/EC50)) was calculated by subtracting the transduction ratio of the agonist (log(Emax/EC50) of the test compound) from the transduction ratio of the reference compound CP55,940 (log(Emax/EC50) of CP55,940 regarding the same pathway. Ligand bias (or bias factor) between two pathways (ΔΔlog(Emax/EC50)) was determined by calculating the difference between the transduction ratio (Δlog(Emax/EC50)) of one pathway and the transduction ratio of the other pathway (Δlog(Emax/EC50) cAMP – Δlog(Emax/EC50) β-arrestin).
ACKNOWLEDGMENTS
Andhika B. Mahardhika, Clara T. Schoeder, and Christa E. Müller gratefully acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG) for the Research Training Group GRK1873 “Pharmacology of 7TM-receptors and downstream signaling pathways," and by the German Federal Ministry of Education and Research (BMBF, BIGS DrugS). Andhika B. Mahardhika was funded by the Ministry of Finance Indonesia in the scheme of the Indonesia Endowment Fund for Education (Lembaga Pengelola Dana Pendidikan [LPDP]). Clara T. Schoeder was supported by a Bayer PhD fellowship. Open Access funding enabled and organized by Projekt DEAL.
CONFLICT OF INTEREST
The authors declare no conflict of interest.

