

Bridging Homogeneous and Heterogeneous Catalysis: Phosphine-Functionalized Metal-Organic Frameworks
Graphical Abstract
Phosphine-functionalized MOFs (P-MOFs) offer novel opportunities in heterogeneous catalysis. In this Minireview, we summarize the synthetic strategies, characterization and catalytic reactions based on the reported P-MOFs. In particular, we remark the current obstacles and possible solutions, including new reaction types and techniques as the perspectives for the development of P-MOF catalysts, highlighting the opportunities behind challenges.

Abstract
Phosphine-functionalized metal-organic frameworks (P-MOFs) as an emerging class of coordination polymers, have provided novel opportunities for the development of heterogeneous catalysts. Yet, compared with the ubiquitous phosphine systems in homogeneous catalysis, heterogenization of phosphines in MOFs is still at its early stage. In this Minireview, we summarize the synthetic strategies, characterization and catalytic reactions based on the P-MOFs reported in literature. In particular, various catalytic reactions are discussed in detail in terms of phosphine ligand structure-function relationship, including the potential obstacles for future development. Finally, we discuss the possible solutions, including new types of reactions and techniques as the perspectives for the development of P-MOF catalysts, highlighting the opportunities and challenges.
1 Introduction
As an emerging class of porous materials, metal-organic Frameworks (MOFs) have been an intensively investigated area of material research over the last decades.1 The high surface area, high crystallinity, structural and functional variety of available organic linkers and inorganic clusters, make MOFs promising candidates for numerous applications, such as gas storage, catalysis and drug delivery. In particular, MOF catalysis has become one of the most productive fields for MOF research where a variety of inspiring results have been reported.2
MOF catalysts can be classified by the type and origin of catalytically active centers as follows, i) single-site MOF catalysts; ii) nanoparticles/nanocluster immobilized MOFs and iii) MOF derived catalysts.3 Among them, single metal based MOF catalysts, due to their highest theoretical atom utilization efficiency and activity, have attracted a considerable attention.4 Compared to cluster supported single metal or MOF mediated single site, MOF supported/encapsulated complexes inspired by homogeneous systems are readily designed on the molecular level and often exhibit higher stability than their homogeneous analogues because of the immobilization and isolation due to pore confinement. One important example are the N-containing ligands (pyridine, bipyridine, porphyrin etc.), where they act as auxiliary ligands for reactions like C−H activation, hydrogenation and etc.5 Moreover, just like homogeneous auxiliary ligands, the functional groups on organic linkers help modulate the microenvironment of catalytic sites through their electronic and steric properties, sieve or enrich substrate molecules at the catalytic sites through interactions with the framework, and lead to great enhancement in activity and selectivity.6
Phosphorus, the number 15 element, lies in the same VA group with Nitrogen and is one of the most widely utilized elements in organometallic chemistry and catalysis. With same valence shell electron configuration, P and N show similar bonding ability and potential valence. Yet, Phosphorus with additional d orbital could act as δ donor and π acceptor, resulting in numerous binding modes and the formation of various phosphorus compounds like phosphide, phosphine, phosphonic acid, etc.7 In general phosphines are considered softer ligands compared to amines and are prepared through nucleophilic substitution reactions, leading to reactive primary phosphines,8 secondary phosphines9 and macrocyclic phosphines.10 Compared with N-containing heterocyclic compounds, both electronic and steric properties of phosphine are modulated through careful substituent design and evaluated quasi-quantitatively through parameters like Tolman Electronic Parameter11 and Tolman Cone Angle.12 Phosphine ligands can bind to almost all metals and are used in catalysis ranging from industrial ethylene oligomerization13 to Nobel Prize winning cross coupling reactions14 and even metal-free organocatalysis.15 However, the abovementioned examples are homogeneous catalyst systems and their application is hampered by challenges associated with their practical utilization such as catalyst separation, recycling and product purification. As such, heterogenization of homogeneous phosphine ligand containing complexes to access immobilized catalysts is a practical and valuable tool for the development of next generation catalysts.
The trail of grafting phosphine complexes onto organic polymers can be traced back to 1978, when polymer immobilization afforded air and moisture stable Pd (0) complexes that could catalyze various coupling reactions. However, the non-porous resins limited the mass transfer and amorphous structure made it difficult to identify the active center for mechanistic study.16 Since then, more porous materials with limited degree of order in the structure such as porous metal phosphonates17 and porous organic polymers18 were reported. Later, with the development of crystalline materials possessing permanent porosity, MOFs have been viewed as a potential solution to the issues stemming from amorphous nature of the support. Their crystallinity allows the precise tuning of the active site through reticular chemistry and postsynthetic modification19 while enabling the rational control of the micro/mesoporosity to overcome mass transfer limitations.20 Moreover, recently more in situ and ex situ characterization methods coupled with theoretical calculations were applied to enhance the understanding and predict the influence of phosphines.21 However, it should be noted that mechanistic study of MOF catalysts is still in its infancy lacking widely accepted theoretical model and operando monitoring methods. As such, combining the mature phosphine-based catalysts and MOF platform to construct new catalysts or immobilized versions of the known homogeneous catalysts is a promising strategy. Immobilized phosphine based catalysts will enable the use of MOFs for asymmetric catalysis and production of value-added chemicals, to widen the area of MOF application.22 The effects of confinement of phosphines and their catalytic properties were discussed in detail in a recent review by Orton et al.23 Kumar et al. published a review focused on the synthesis and the application of Phosphorus-containing POPs in catalysis and organic synthesis.24 The synthetic challenges of phosphine linkers have been reviewed by Gäumann et al.18 This review will discuss the successful examples of combining strength of homogeneous phosphine catalysis with MOF supports to catalyze a variety of reactions. In addition, new development directions and potential applications are forecast.
The contributions listed above have thoroughly covered the synthetic and structural aspects of Phosphorus-containing porous materials like metal-organic framework/cage, covalent organic frameworks and porous organic polymers. As such, in this review we focus on the similarities and differences between homogeneous and heterogeneous catalysis by molecular complexes of phosphorus. Characterization techniques as well as catalytic applications will be introduced combining the strength of phosphine complexes and MOFs. Unmet challenges and possible solutions that open new horizons for the MOF-based catalyst research are also discussed.
2 Synthesis and Characterization of Phosphine-Functionalized MOFs
This section covers the synthetic strategies as well as characterization techniques of P-MOFs. Depending on the targeted application and function, the P-MOFs can be classified into two groups, MOFs with structural phosphines and MOFs with functional phosphines. In the MOFs with structural phosphines the phosphines coordinate to metal clusters to create the MOF framework and open phosphine sites are not available for further coordination by metals to generate single-site catalysts. Whereas in MOFs with functional phosphines, the key feature is the open phosphine coordination sites, that are able to coordinate to transition metals and form single-molecular catalytic sites.
Thus, the construction of P-MOFs, based on phosphine type, can be divided into three methods: direct synthesis with structural and functional phosphines, and ligand exchange (Scheme 1). Postsynthetic modification of MOFs with structural phosphines, causes collapse of MOF structure and is not a viable method for preparing P-MOFs. The first two methods include (Scheme 1a, b) synthesis of P-MOFs from phosphine containing linkers and metal precursors while the third method involves postsynthetic decoration or immobilization of phosphines on MOF crystals (Scheme 1c). Below we compare the strengths and shortcomings of the methods of P-MOF preparation from the perspective of heterogeneous catalysis We do not cover the details of ligand synthesis as it was reviewed elsewhere.18

Schematic representation of ways to build P-MOFs for catalysis. Direct synthesis using a) structural phosphine, b) functional phosphine and c) Postsynthetic modification. Green: metal cluster, Orange: reactive metal site, Grey: organic ligand, Purple: phosphine.
2.1 Direct Synthesis of P-MOFs
In the early stages of research, the P-MOFs were prepared from phosphine containing linkers obtained from simple structural transformation by adding proper binding sites to enable metal binding required to form the MOF scaffold. For example, adding trans carboxylates to triphenyl phosphine, where the P atom acts as tritopic node leading to a trigonal-pyramidal linkers.25 Compared with traditional planar triangular linkers, the pyramidal geometry enables the fabrication of novel MOF topologies. The exposed phosphine influences gas adsorption properties of Lewis acidic CO2.26 With the ability for coordination with a variety of metals, phosphines have been viewed as building blocks for organometallic self-assemblies (ring, cage, polymer etc.) even before P-MOFs became widely acknowledged.27 Puddephatt,28 James,29 Humphrey,25 Lin,30 and Cohen31 have pioneered the synthesis of P-MOFs with phosphines coordinated with metal nodes, leading to low-valent metal complexes. For example, recently, Cohen's group reported a series of MOFs with low-valent (0,1) metal nodes connected by phosphine ligands, resulting in novel structural and luminescent properties.32 However, only a handful of low-valent P-MOFs were reported, possibly due to the difficulty in phosphine ligand synthesis. The coordination polymers based on phosphine-low valent metal coordination were reviewed by Cohen et al. in the recent minireview.33
Early progress has also been made using phosphonic acid or phosphonates as binding sites due to their strong binding ability to metals.34 Interestingly, unlike the monodentate phosphine-metal coordination, the phosphonate with CPO3 tetrahedral geometry could coordinate with multiple metals through coordinating or bridging oxygen actoms. Other than the unique coordination mode, the stronger metal phosphonate interaction endows MOFs obtained in this fashion with high thermal and hydrolytic stability. However, in metal phosphonate derived MOFs, the phosphorus functionality is engaged in binding with oxygen or metal node, leaving no room for anchoring of catalytically active moieties or substrates. Thus, they are not included in the discussion of P-MOF based catalysts. In contrast, nonstructural phosphines with free binding sites are preferred in MOF catalysts design.
Phosphines linked with amines lead to functionalized amino phosphines which are widely applied in the design of ethylene oligomerization catalysts.13 Through manipulating the substituents on phosphine, the selectivity and activity of complexes are well optimized.35 This strategy of ligand tuning has been applied in homogeneous phosphine ligand design extensively. For example, Kempe and co-workers have developed a combinatorial library strategy for parallel synthesis of pyridine-amino phosphines for catalyzing Suzuki coupling reaction.36 In MOF catalysts design, in addition to optimization of electronic and steric properties, structural modification of phosphines also lead to microenvironment tuning.37
A promising method for introducing phosphine functionality into MOFs is through functionalizing the common linear terephthalic acid (H2BDC) linker with a phosphine moiety (P1 and P2, Figure 1).38 MOFs synthesized from terephthalic acid like MOF-5, UiO-66 and MIL-101 can be functionalized with these phosphine containing linkers. It is noteworthy to mention that MOF versions with expanded pores is achieved using extended version of these linkers.26 New P-MOFs can be prepared with this strategy so long as the ligands can withstand the hydrothermal synthesis conditions. For, example, P-MOFs with stable bis(phosphine) or PCP pincer functionalized linkers were prepared with this strategy (P8 and P9, Figure 1).39 The examples of phosphine ligands with different connections are divided based on structural and functional phosphines, as summarized in Figure 1 (P1 to P11). It is worth mentioning that P center in structural phosphines, though able to coordinate with the MOF metal nodes as well, will not influence the connectivity. Because the soft PR3 is incompatible with metal clusters, especially the hard early transition metals that are commonly used to build stable MOFs (Zr4+, Fe3+).40 This feature guarantees that fabrication of P-MOFs is not influenced by coordination to metal precursors in the MOF scaffold, both in direct synthesis and postsynthesis methodologies. Usually, ligands or metal clusters with higher connectivity form MOFs with higher stability. This concept is applied in robust P-MOF catalyst design in the following sections.

Typical examples of 2,3 and 4-connected phosphine ligands used to build P-MOFs. Functional phosphines: green. Structural phosphines: magenta.
2.2 Postsynthetic Modification Method
The abovementioned presynthesis of P-MOFs often requires relatively harsh hydrothermal conditions and sets high standards on the stability of phosphine ligands. As a result, postsynthetic modification (PSM) method, including postsynthetic metalation and postsynthetic linker exchange, is viewed as a powerful tool to decorate and functionalize the pore environment under mild conditions. It is noteworthy that PSM generally inherits the geometry and topology of precursor MOFs undergoing ligand exchange, and the precursor MOFs should be chosen according to the requirements of the reaction.
Derived from the traditional triarylphosphine, tritopic phosphine tricarboxylate ligand provides ideal Lewis basic sites for the coordination of metal. In 2011 Humphrey group reported the first example of postsynthetic metalation on P-MOF by anchoring Au(I) on the tris(p-carboxylated) triphenylphosphine linker (P6, Figure 1).41 The anchored Au(I) sites coordinated with the C=C bond in 1-hexene, which caused a strong desorption hysteresis, indicating that the phosphine-complex participates in the host-guest interaction and has potential for catalytic activation of Lewis basic substrates. Later, the same group reported the postsynthetic methylation of the PCP pincer-Pd complex to facilitate the adsorption of CO2.42 The postsynthetic metalation enables not only change in pore structure but also construction of chiral centers for asymmetric catalysis. Falkowski et al. used BINAP derived carboxylate linkers (analogue of P4, Figure 1) and Zr6 clusters with UiO-66-type topology suitable for facile postsynthetic metalation.43 This opened up a new avenue for creating chiral center for asymmetric catalysis, which will be discussed in detail in the next section.
The prerequisite for postsynthetic introduction of phosphine groups is the preservation of MOF scaffold, which can only be produced with non-structural phosphine functionalities. In 2012 Gascon group reported a chloromethylation method, in which the benzyl chloride functionalized MOF underwent a nucleophilic substitution with LiPPh2 to afford benzyl phosphine immobilized MOF.44 The substitution induced phosphine exchange can also happen on more common amino group. Castellanos et al. reported in situ reaction between amino group and diphenylphosphinic chloride to attach O=P-NH(Ph2) functionality within MIL-53(Al). The introduction of diphenylphosphinyl groups was determined to alter the luminescent properties through changing energy levels of linkers.45 It is noteworthy that the results above are both based on MIL-53, which has large one-dimensional channel and robust framework stable under post-synthesis modification conditions.
Postsynthetic ligand insertion/exchange is an alternative and practical way to introduce phosphine groups, which avoids the possible destruction of the framework. Humphrey46 and Zhang47 applied postsynthetic ligand insertion on stable MOF-808 for heterogeneous bifunctional catalysis and uranium adsorption, respectively. It is generally believed that the Zr/Hf clusters in MOF-808 provide robust yet modifiable support with accessible formate bound sites amenable to exchange by carboxylate or phosphoric acid. Zhang group synthesized (2-(diethylphosphoryl)ethyl)phosphonic acid (DEPA) and grafted it to the 1D channel of MOF-808 and NU-1000 to construct MOF-808-PO and NU-1000-PO, which revealed the generality of this method (Figure 2). This modulator-exchange method is possible without the loss of crystallinity or porosity, with partial occupation of void space by bulky phosphine oxide ligand. Furthermore, the phosphine oxide could be reduced to phosphine that can generate catalytic complexes with transition metals. Recently we used the ligand exchange method to tether the PPh2 group on the surface of UiO-66. Compared with other phosphine modification methods, the ligand exchange can be applied for a variety of MOF scaffolds, providing a greater freedom in the choice of MOF candidates, and isolated active sites through immobilization on MOF surface. Because of the mild conditions of the linker exchange, the PSM method also permits modification of electronic and steric properties of the immobilized functionality, such as varying the substituents on the phosphine ligand, thus enabling the fine tuning of the catalyst system. These strategies have enriched the P-MOF database and expanded the toolbox for functionalizing MOFs with phosphines. The P-MOF catalytic examples will be discussed in detail in Section 3.

a) Synthesis of (2-(diethylphosphoryl)ethyl)phosphonic acid (DEPA). Schematic representation of postsynthetic modification to construct b) MOF-808-PO and c) NU-1000-PO. Reproduced with permission from Ref. [47]. Copyright 2021 Royal Society of Chemistry.
2.3 Characterization
In the spirit of bridging homogeneous and heterogeneous catalysis, characterization methods common in both homogeneous and heterogeneous systems will be discussed in this section. Spectroscopic characterizations are widely used in catalyst study, especially nuclear magnetic resonance (NMR), X-ray absorption spectroscopy (XAS) and Infrared spectroscopy.
Most widely applied identification technique for P-MOFs is the solid-state nuclear magnetic resonance (SS NMR), which provides non-interference structural information on the immobilized phosphorus functionality. For example, Castellanos et al. used solid-state 31P NMR spectroscopy to confirm the successful formation of the phosphinic amide after the PSM.45 The spectrum of PO-NH-MIL-53(Al) consists of three peaks, two minor ones at δ=30.0 and 24.2 ppm and a more intense one at δ=10.2 ppm. Compared with homogeneous dimethyl linker, the immobilization of ligands in MOF shifts the peaks slightly upfield, while the third peak was ascribed to diphenylphosphinic acid residue. Specific NMR methods can give more structural information like the relative distance of functional groups, which is not easily measured in the free-standing homogeneous complexes. Deng group reported the construction of a “molecular vise” (MV) by pairing a tritopic triphenyl phosphorus(III) linker (P6, Figure 1) and a monotopic linker in opposite positions within a MOF.48 The monotopic linker points to the open phosphine site, where the distance between them can be controlled through changing monodentate linkers (Figure 3a). Quantification of the distance was built on the correlation between distance of linkers and the “decay curve” measured from 1H-31P rotational echo double-resonance (REDOR) SS NMR (Figure 3b). Generally, closer distance within selected pairs results in rapid decay, where the Y axis is calculated from the specific value of intensity of peaks with (S) without (S0) 1H-31P REDOR dipolar dephasing (Figure 3c). Thus, with the help of simulated structural model, the distance was estimated and then confirmed experimentally with the precision that cannot be achieved by other spectroscopic methods. Then, the distance between monotopic linkers and phosphine were determined for the sample containing dichloroacetic acid (DCA) linker.

a) Schematic representation of “Molecular Vise” with Dichloride acetic acid (DCA) as ligand. b) 1H-31P REDOR for P-MV-DCA c) dipolar dephasing of REDOR peak at 5.3 ppm for P-MV-DCA. Reproduced with permission from Ref. [48]. Copyright 2020 American Chemical Society.
The utilization of P K-edge XAS to study MOF-immobilized phosphorus functional groups was demonstrated as early as 2015, where Bokhoven group implemented it to determine the ratio between phosphine and phosphine oxide.49 Two P-MOFs with MIL-101 topology were synthesized through traditional solvothermal method using 2-diphenylphosphinoterephthalic acid (BDC-PPh2) (P1, Figure 1). Then K-edge XAS of phosphorus was measured for MOFs and compared with BDC-PPh2 and oxidized BDC-POPh2. X-ray absorption near-edge structure (XANES) is sensitive to the valence of elements and unlike SS NMR can be used for samples containing paramagnetic nuclei. This straightforward method gave out clear information of the components of MOFs without the need for computation. Furthermore, with the help of structure simulation, more information including coordination number, distance and oxidation state can be determined using XAS.
Due to the insoluble nature of P-MOFs, traditional liquid phase characterization methods are not viable for intact MOF samples. To thoroughly analyze and understand the structure of materials, a combination of multiple instrumental techniques is usually applied; X-ray diffraction (XRD) is used to determine the crystalline structure, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) are applied for morphology detection and X-ray photoelectron spectroscopy (XPS) to determine the valance state of elements. However, to study the mechanisms of MOF catalyzed reactions, more nondestructive and effective in situ characterization techniques are necessary.50
3 Examples of Reactions Catalyzed by P-MOFs
With the help of mature pre/postsynthetic functionalization methods and new structural characterization techniques, P-MOFs have been widely applied in heterogeneous catalysis. Not only the heterogeneous versions of reactions catalyzed by homogeneous phosphine complexes were reported, but also the ones like photocatalytic reactions and CO2 reduction not very common for homogeneous phosphine complexes started to emerge. The reactions catalyzed by P-MOFs are summarized in Table 1, with the catalytically active site and MOF support platform. In this section, we attempt to critically review the examples of catalysts based on P-MOFs and identify the possible future directions to address the unmet challenges with the help of MOF catalysts.
P-MOFs (Conventional name or CCDC number) |
Active sites |
Reactions |
Ref. |
|---|---|---|---|
BINAP-MOF (UiO-66) |
Rh |
Asymmetric addition |
[43] |
LSK-3 (IRMOF-9) |
Phosphine |
Knoevenagel condensation, Cycloaddition |
[51] |
P1-MOF (1485918) |
Rh |
Hydrosilylation |
[40] |
P1-MOF (1485918) |
Rh |
Hydrogenation |
[40] |
PCM-101 (1581200) |
Au |
Alkyne hydroaddition |
[46] |
Zr12-P-MOF(1585874) |
Phosphine |
CO2 cycloaddition |
[52] |
PCM-107 (1913704) |
AuCl |
Alkyne hydroaddition |
[53] |
P1-MOF (1485919) |
Ir |
C−H Borylation |
[40] |
SulP1-MOF-808 (Hf)-Ir (MOF-808) |
Ir/Rh |
reductive amination |
[54] |
Pd-1@Ni-MOF ([Ni8(OH)4(H2O)2(L)6]n) |
Pd |
Heck Arylaiton |
[55] |
Zr-P-Ir (as P1-MOF) |
Ir |
C−H Borylation |
[56] |
MOF-P-Co (as P1-MOF) |
Co |
C−H Borylation |
[57] |
Pd-LSK-15 (MIL-101Al) |
Pd |
Suzuki Coupling |
[58] |
Pd/MIL-101-PPh3 (MIL-101) |
Pd NPs |
Suzuki and Heck Coupling |
[59] |
Hf-Ru-Au (Zr-BTB) |
Au |
Photocatalytic C−C coupling |
[60] |
Ru-LSK-15 (MIL-101) |
Ru |
Formic acid dehydrogenation |
[61] |
[Cu(C14H12O6.5P2)(4,4′-bipy)0.5(H2O)] (1872673) |
Photogenerated electron |
Reduction of Cr(VI) to Cr(III) |
[62] |
3.1 Addition Reactions
Addition reactions include various reaction types like hydrogenation, hydrosilylation, cycloaddition, hydroaddition and asymmetric catalysis. As one of the most explored reactions in organic catalysis, it was mostly reported in the early stages of P-MOF catalyst design, which was done in the spirit of immobilizing analogues of transition metal phosphine complexes on robust heterogeneous MOF supports. 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) was first selected as parent ligand due to its high activity and special chirality, which ensured successful transfer of chirality to the P-MOF. Wenbin Lin group has produced some pioneering work with heterogenization of BINAP complexes, where they upgraded the support from disordered zirconium phosphonates to MOFs.17 In 2014, Lin group reported a chiral MOF with fcu topology like UiO-66 with Zr6 cluster and dicarboxylic acid linker derived from BINAP (Figure 4a).43 Immobilized chiral catalysts were obtained through postsynthetic metalation with Rh and Ru (Figure 4b). As proof-of-concept P-MOF catalyst, BINAP-MOF-Rh is capable of catalyzing asymmetric additions of arylboronic acids to 2-cyclohexenone with 80–99 % isolated yields and >99 % ee, which is similar in selectivity but more active than its homogeneous counterpart BINAP-Rh. The high reactivity was ascribed to site isolation of chiral centers, preventing intermolecular deactivation pathways. In 2019, Jiang group worked with Yong Cui group to report 3 Indium MOFs63 with chiral phosphoric acid center in the tetracarboxylate linkers. The chiral phosphoric acid was applied in asymmetric addition/condensation of amine with aldehyde/ketone, which required presence of a Brønsted acid. Moreover, the steric effect around chiral center dominates the performance of asymmetric catalysis, where the percentage of sterically bulky anthracenyl groups is proportional to the ee value. Recently, Cui group used the PSM approach to acquire chiral Zr-MOF.64 In this case, the step-by-step formation of chiral Rh-monophosphorus complex prevents the deactivation caused by dimerization (P10, Figure 1). The catalytic asymmetric hydrogenations of enamides and α-dehydroamino acid esters proceeded with excellent yields and ee values (both up to 99 %). The significantly enhanced activity of Rh-monophosphorus species within P-MOF is ascribed to the presence of an open coordination site in contrast with homogeneous Rh-bisphosphorus counterpart. The abovementioned reports have demonstrated the strength of asymmetric phosphine ligand as chiral center and MOF scaffold as robust support. However, the potential of MOF as a highly tunable and ordered material is underexplored. Through careful modulation of the chemical/spatial microenvironment of chiral centers, additional chemoselectivity/regioselectivity is demonstrated by P-MOF catalysts.65 Bokhoven group reported a MOF-5 isoreticular structure based on diphenyl-phosphine tethered BPDC and Zn.51 The afforded PPh2 groups within the pore act as organocatalyst and were screened for coumarin synthesis and Knoevenagel condensation. Yet, the catalysts were not reactive for umpolung addition and [3+2] cycloaddition. This alternating activity was determined to be influenced by steric hindrance of the framework through molecular modeling and intermediate determination. Because the reactions start with nucleophilic addition of phosphine, forming the zwitterionic intermediate in which the ethyl 2,3-butanedienoate substrate was impeded by the framework, leading to much higher energy barrier. This demonstrates that, the “environment” of catalyst greatly influences the activity and selectivity of reactions. And this “environment” are the substituents in homogeneous systems and pore structure and substituents in P-MOFs.

a) Synthesis of BINAP-derived dicarboxylic acid, H2L. and b) Postsynthetic metalation of BINAP-MOF for asymmetric addition and hydrogenation. Reproduced with permission from Ref. [43] Copyright 2014 American Chemical Society.
In contrast to bidentate linkers, triarylphosphine-derived carboxylate linkers help build new topologies and novel geometries of MOFs. In 2016, Lin group reported the first example using MOF scaffold to stabilize the mono(phosphine)-metal complexes, where the triarylphosphine-derived tricarboxylate anchored Rh/Ir monophosphine complexes catalyze reactions such as hydrogenation of alkenes, hydrosilylation of ketones and alkenes, and C−H borylation.40 The afforded catalysts show superior activity to homogeneous counterparts due to the confinement effect on mono(phosphine)-complexes provided by MOF. In addition, new coordination geometry could be achieved in the limited pore-space of MOF. Humphrey group synthesized the first example of trans-phosphine-metal complex within a MOF pocket.46 Two triphenylphosphine-derived carboxylate linkers connected by trinuclear clusters and pyridine-based pillars, forming phosphine complexes through postsynthetic metalation. Coordination of AuCl to each of the trans-phosphine binding sites, forces a large distortion of the parent framework due to formation of two Au-monophosphine complexes. In contrast, two CuBr molecules coordinate directly between the trans-P sites to form dimers that mimic solution-phase complexes, but that are geometrically strained due to steric constraint exerted by the MOF scaffold. The constrained metal phosphine complexes are active in heterogeneous hydroaddition reactions, which shows different product selectivity and higher activity compared to the homogeneous analogue due to the distorted geometry of the complexes. Fabrication of novel P-MOFs could also go through ligand exchange. Ditzel et al. demonstrated the facile synthesis of two MOFs from carboxylate bridged Zr12 clusters, in which exchange to tritopic phosphine linker results in MOFs with novel kgd topology.52 Preliminary tests indicate that it selectively catalyzes the coupling of CO2 and styrene oxide to styrene carbonate in the presence of [nBu4N]Br as co-catalyst. In 2019, Humphrey used one-pot synthesis of poly(carboxylated) aryl(phosphine) ligands and mixtures of Co(II) and Au(I) precursors to form P-MOF and metal-phosphine complexes at the same time.53 The resulting PCM-107 is active for alkyne hydroaddition, while PCM-74 bears the novel trans Cu2I2 complexes.
3.2 Coupling Reactions
In the early stages of development of MOF-based catalysts the coupling reactions, have received less attention than addition reactions. This is due to the factors associated with coupling reaction conditions and preparation of the MOF catalysts for coupling since stable MOF scaffold is necessary to withstand the basic conditions required for cross coupling reactions; and a confined space or stable P-M complexes are needed to prevent the demetallation during catalytic cycle. Instead, nitrogen-containing ligands with higher base stability, especially bipyridine/ phenanthroline functionalized MOFs, flourished as chelating and site isolation ligands.66 With the development of new synthetic strategies for preparing P-MOF based catalysts, several examples were published recently. In 2019, Trzeciak et al. have immobilized two PdCl2P2 type palladium complexes of [P=tri(1-piperidinyl)phosphine and triphenylphosphine] at Ni-MOF via confining the Pd complexes within the pores to catalyze Heck arylation of estragole by iodobenzene to the corresponding stilbene with high efficiency.55 The afforded catalysts show size selective low activity toward bulky eugenol. Last year, two reports were published by Ranocchiari58 and Ren59 for highly active Suzuki and Heck coupling reactions (Table 2). It is noteworthy that MIL-101 Al/Cr was used as catalyst support in both cases due to its high stability and porosity. Single Pd atoms were stabilized by monophosphine ligand at active sites. Notably, Pd-LSK-15 possesses comparable reactivity with homogeneous Pd(PPh3)4 but lower recyclability than Pd single atom supported on MOF or polymers (Table 2). These reports have demonstrated the utility of phosphine ligands for anchoring transition metal complexes.
|
|||||
|
|||||
|---|---|---|---|---|---|
Catalyst |
Solvent |
Base |
Yield (%) |
Reused(Yield)[c] |
Ref. |
Pd/MIL-101-PPh3 |
EtOH/H2O |
K2CO3 |
99 |
5(89) |
[59] |
Pd-LSK-15 |
toluene/H2O |
Et3N |
65 |
3(37) |
[58] |
Pd(PPh3)4 |
toluene/H2O |
Et3N |
66 |
NA |
[58] |
Pd/CPPa |
EtOH/H2O |
K3PO4 |
99 |
5(92) |
[67] |
Pd/POL-PPh3[b] |
toluene/H2O |
K3PO4 |
99 |
10(96) |
[68] |
- [a] Pd ions anchored on phenanthroline based conjugated porous polymer (CPP). [b] Pd loaded porous polymer based on PPh3. [c] Recycled runs (yield of last run are in parentheses).
Moreover, through rational design and arrangement of linkers, the metal-phosphine complexes were assembled with photosensitizers, affording unprecedented photoredox reactions. Lin group reported a metal-organic layer (MOL), Hf-Ru-Au, with Ru(bipyridine)32+-type as photosensitizers and (phosphine)-AuCl as active site (Figure 5a, b).60 Inspired by homogeneous systems, it is the first reported P-MOF photocatalyst for cross-coupling of allenoates, alkenes, and alkynes with aryldiazonium salts.69 Thanks to the closely assembled photosensitizer Ru-PS and monophosphine-Au(I) complex, the transfer of electron and radical intermediates are greatly promoted, showing similar yield with much lower loading of Ru and Au compared to homogeneous counterpart (around 14–200 times less). Furthermore, site isolation of (phosphine)-AuCl complexes in Hf-Ru-Au prevents Au catalyst deactivation via ligand redistribution, Au(I) disproportionation, and aryl-phosphine reductive elimination, affording high recyclability and TON (Figure 5c).

a) Schematic synthesis of Hf-Ru-Au. b) Stick model showing close attachment and site isolation of P-AuCl with Ru-PS. c) Cross-coupling of allenoates, alkenes, and alkynes with aryldiazonium salts (a: Isolated yields and TONs are shown in parentheses). Reproduced with permission from Ref. [60]. Copyright 2022 American Chemical Society.
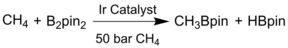
Underligated mono(phosphine)-M coordinated complexes are well-known to be highly active for cross coupling reactions that include changes in valence, coordination number of active sites and the chemical environment over the course of the reaction.70 Clarke et al. reported the C−N bond formation through reductive amination reaction.54 The sulfonated phosphines exchanged the formate group on the Hf6 cluster of the robust and stable MOF-808 and were metalated with [IrCl(cod)]2 or [Rh(acac)(CO)2] to stabilize the immobilized single molecular monophosphine complex. Compared with homogeneous complexes which produce partial reduction product with low recyclability, prepared MOF catalysts show good conversion (96 %) and selectivity towards secondary amine (99 %) over up to 5 runs. This work represents the potential of Zr/Hf based P-MOFs with Lewis acidic sites and phosphine-metal complexes for bifunctional tandem catalysis. Formation of C−X bond (X=B, N, S) through direct C−H activation is a simple and atom-economical method of functionalization. In 2019, Lin group used tris(4-carboxylphenyl)phosphine ligands to synthesize an effective P-MOF catalyst for methane borylation.56 The low-coordinated mono(phosphine)-Ir complex stabilized in MOF activated methane to get CH3Bpin (pin=pinacolate) with a turnover number (TON) of 127 at 110 °C, which is comparable to the state-of-the-art homogeneous and silica supported Ir catalyst, showing potential for the activation of inert C−H bonds (Table 3). Density functional theory calculations revealed that the catalytic cycle starts from the four-coordinate (P1)IrIII(Bpin)3 and then goes through oxidative addition of methane to form the six-coordinate (P1)IrV(Bpin)3(CH3)(H) intermediate. Thus, compared with other bidentate Ir complexes with chelating phenanthroline, bipyridine, and bisphosphine ligands forming sterically hindered seven-coordinate intermediates, the activation barrier is reduced for mono(phosphine)-Ir catalyst. This mediation effect of low-coordinate metal, which often requires bulky substituents and specific ligand design in homogeneous organic catalysis, is easily achieved in P-MOFs due to the spatial confinement effect. Later, Manna et al. used the extended version of tri(aryl)phosphine derived linker to build P-MOF, stabilizing monoligated P-Co complex for C−H borylation and alkene hydroboration.57 The afforded MOF-P-Co could catalyze ortho-xylene borylation affording meta-Bpin with yield up to 86 % and TON up to 30,000, indicating superior stability than homogeneous metal or metal free catalysts.71 The high activity was also ascribed to the low-coordinate intermediate, in this case, four-coordinated MOF-P-Co(H)(Bpin)2. In contrast, the homogeneous control (Ph3P-Co), even added in 1 : 1 molar ratio, forms (Ph3P)2CoCl2 and CoCl2 through Schlenk-type equilibrium.
|
|||||
|
|||||
|---|---|---|---|---|---|
Catalyst |
Solvent[a] |
Temp. (°C) |
Yield (%) |
TON |
Ref. |
Zr-P1-Ir |
CyH |
110 |
38.0 |
127 |
[56] |
UiO-67-Ir[b] |
CyH |
150 |
27.8 |
56 |
[72] |
IrP-SiO2[c] |
CyH |
150 |
82.5 |
1217 |
[73] |
Ir-dmpe[d] |
CyO |
150 |
31 |
170 |
[74] |
- [a] CyH=cyclohexane, CyO=cyclooctane. [b] Ir catalyst supported by UiO-67-phenanthroline(phen). [c] Ir dmpe on SiO2, dmpe=1,2-bis(dimethylphosphino)-ethane. [d] Homogeneous (dmpe)Ir(COD)Cl, COD=1,5-cyclooctadiene.
3.3 Other Reactions
Besides organic small molecule transformation, more gas/vapor phase reactions highlighting the ability of MOF based catalysts to operate under gas phase conditions have gradually been reported. In 2015, Bokhoven et al. reported simple strategy of anchoring a molecular ruthenium complex in a phosphine-functionalized MOF (MIL-101-PPh2).61 The afforded catalyst is highly active for dehydrogenation of formic acid, and exclusively produces hydrogen and carbon dioxide with turnover frequency of around 2300. This state-of-the-art catalyst has shown the strength of P-MOFs in bridging homogeneous and heterogeneous catalysis, where recyclability and stability of the Ru complex was greatly enhanced while maintaining the activity.
As for new reaction types, due to the Lewis basic nature of phosphine ligands, combining the bulky Lewis base with Lewis acid to form the frustrated Lewis pair (FLP), is a well-known strategy to activate small molecules via the concerted action of Lewis acid and base. As a result, this strategy is well-established in homogeneous systems using sterically impeded phosphine-borane pairs to heterolyze H2 and subsequently engage in hydrogenation. Traditional P-B pairs are reported in Figure 6.75 Porous materials (MOF, COF, porous polymer) with inherent cavity to trap the FLPs are ideal catalysts supports. The current progress and challenges in efficient transformations using porous FLP catalysts in advanced catalysis was recently reviewed by Ma..76 Usually, one end of FLPs (phosphine or borane) was heterogenized in porous materials as binding sites, while the other group was trapped in the pore to create specific environment for selective hydrogenation reaction.

Examples of H2 activation using P-B frustrated Lewis pairs. Reproduced with permission from Ref. [75]. Copyright 2015 American Chemical Society.
4 Summary and Perspectives
The past decades have witnessed tremendous achievement made in the field of MOF heterogeneous catalysts. The development of P-MOFs, by contrast, is highly underexplored. In this section we summarize the complicated reasons for the limited development of P-MOFs and possible solutions. Then we forecast the future direction of P-MOFs based on the cutting-edge results in the fields of both homogeneous and heterogeneous catalysis.
4.1 Limitations and Possible Solutions
The first limitation of P-MOFs is the intrinsic structural bulkiness of phosphine ligands. Compared with bipyridine based ligands which can easily exchange biphenyl groups in the MOF structure, the triangular pyramidal phosphines are too bulky to fit in the pores of most microporous MOFs. Because of this, direct synthesis with phosphine linker is required for even distribution of ligands, while PSM method will graft phosphine ligands on the surface.38 On the other hand, the special non-planar structure of phosphine promotes the development of new MOFs. Recently, Wang group built the metal-organic layers (MOL) with trigonal pyramidal phosphine oxide ligand.77 In contrast to other MOLs, the Hf6 oxo clusters were alternately connected with Hf12 oxo clusters, resulting in wavy 2D structure (Figure 7). Similar topology could be achieved with reduced tris(4-carboxylphenyl)phosphine (TPP) ligand, which provide metal coordination sites for catalysis. As such, structure of phosphine incorporated linkers could enable preparation of novel P-MOFs.

Structural model of Hf-TPO-MOL. (a) Hf6 and Hf12 SBUs and the TPO ligand; (b, c) Structure of Hf-TPO-MOL viewed in different directions; (d) Formation of the 3,6-connected 2D kgd lattice. Reproduced with permission from Ref. [77]. Copyright 2022 Royal Society of Chemistry
Secondly, the stability and recyclability issue commonly observed in MOF catalysts but more severe with electron rich phosphine functionalities due to the oxygen sensitivity. As a result, relatively stable triphenylphosphine (PPh3) derivative-based linkers are used for the functionalization of MOFs, which was discussed in the Section 2.1. Also, synthesis of P-MOFs through PSM rather than direct synthesis will help minimize the possibility of partial oxidation during solvothermal synthetic conditions. Generally, the reactive functionalities on organic linkers like amino and chlorine substituents are used to tether phosphine ligands to MOF structure through imine condensation or cross coupling reactions.18, 45 We further suggest that other post-reaction characterizations like gas adsorption, Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES), and SS NMR are performed to determine the change in amount and state of active sites, especially for some reaction pathways where redox and demetallation happen.
Finally, compared with the homogeneous phosphine complexes, in situ characterization methods of P-MOFs are limited due to their solid nature. Especially, lacking in situ monitoring of structural transformation of catalysts causes great difficulty in mechanistic studies that would enable a deeper understanding of factors that control catalyst activity and selectivity. Homogeneous analogues of the MOF-immobilized catalysts can be viewed as a model; however, MOF catalysts usually display contrasting activities and selectivities and mechanistic insight into the reasons for these differences would be invaluable. To monitor the catalytic transformation which mostly happens in liquid phase, an in situ characterization method that bridges the solid catalysts with solution-state environment is needed. In 2020, our group reported an oligomer-immobilization method to solubilize and disperse the MOF nanoparticles in liquid phase.50a Phosphonic acid was selected as binding group on the oligomer, which could be grafted on metal clusters on MOF surface. Furthermore, as a non-destructive and non-interfering method, transmission infrared (IR) spectroscopy was applied on solubilized MOF particles. Photochemistry of the bipyridine-Re(CO)3Cl complex grafted on MOF was monitored, which mimics the homogeneous molecular catalyst. Recently, our group has expanded this strategy towards electrochemical reactions catalyzed by MOF-immobilized molecular catalysts.50b In this work, the solubilized MOF catalysts were applied in CO2 electroreduction and its reaction intermediates were studied using Infrared Spectroelectrochemistry (IR-SEC). The structure of captured intermediate was then compared to that of its homogeneous counterpart. It is demonstrated that, due to the MOF immobilization, no dimeric intermediate was found among the intermediary complexes. Overall, developing more in situ/operando characterization techniques will not only deepen our understanding of the mechanism of P-MOF catalysts but accelerate the development in the whole of heterogeneous catalysis field.
4.2 Perspectives
Research on P-MOFs, as a venue for bridging homogeneous and heterogeneous catalysis, can take inspiration from the mature yet still flourishing research on phosphine complexes. In the long run, two important aspects should be considered: 1) deepening the understanding of structure-function relationship to enhance the activity and selectivity of existing reactions; 2) widening the substrate scope for the known reactions or explore new reaction types. For example, Suzuki–Miyaura cross-coupling reaction, ever since its invention, employed mostly triarylphosphine ligands. Later, researchers optimized the structure of the phosphine ligands, and determined that the bulky and electron-rich phosphine ligands greatly enhance the activity of the catalyst. The complexes of these bulky ligands were subsequently, applied to catalyze other coupling reactions, such as C−N, C−O and C−S bond formation.78 Similarly, optimizing the coordination environment of active site, through both steric and electronic effect, is accomplished through alternating substituents on phosphines. This is in line with the modulation of “microenvironment of active site”, which determines the reactivity and selectivity of reactions.23
Moreover, with the implications of new and rapidly changing computational chemistry, development of new reactions will not be limited by experimental conditions. In 2019, Jiang group used theoretical calculation to “create” two new chiral MOFs with fcu and bct topology based on dicarboxylate linker 4,4′-biphenyldicarboxylate (BPDC)-phosphoric acid.79 Then Gibbs free energy of asymmetric transfer hydrogenation of imines was calculated as indicator of reactivity. This work has shown us the power and potential of pure theoretical simulation in developing P-MOF catalysts. In homogeneous phosphine study some outstanding work using machine learning to predict the catalytic properties have been reported. Schoenebeck et al. reported an unsupervised machine learning workflow that uses only five experimental data points to predict the possible formation of dinuclear Pd(I) phosphine complexes.80 In the same year, Doyle group used the powerful machine learning strategy to evaluate the performance of mono(phosphine)-Ni/Pd catalysts, where minimum percent buried volume [% Vbur (min)] was calculated and used as descriptor.81 Compared with other steric descriptors like cone angle, % Vbur (min) is more indicative as it focuses on the chemical environment close to the P atom, what we call “first sphere” effect. Interestingly, a clear watershed between 30–35 % can be found when plotting % Vbur (min) vs. yields of catalyst. That is to say, the % Vbur (min) can be viewed as the threshold to predict the performance of C−C cross coupling reactions, which may vary based on metal and halide.
For future development of P-MOFs in heterogeneous catalysis, maximizing the advantages of active phosphine ligands while coupling the high crystallinity and tunability of MOFs will be the ideal strategy. In this regard, the homogeneous phosphine ligand systems will be viewed as “guidebook” rather than “template”. The chemical environment of tethered phosphine within MOF pore is different from the free solution in homogeneous systems, which will be both challenge and opportunity in catalyst design. And the performance of the resulting catalyst, often comparable to its homogeneous counterpart, can be correlated to a set of defined parameters, such as site-isolation effect inhibiting deactivation, spatial control of adjacent active sites and substrate enrichment in pore space. Therefore, there is an exciting opportunity in heterogenizing molecular catalysts into P-MOFs through creative design.
In this minireview, we have highlighted the synthesis, characterization, and application of phosphine decorated MOFs with examples from the literature. Despite of the remarkable progress that has been made by MOF catalysts and the relative maturity of homogeneous phosphine systems, P-MOFs have yet to be exploited to their full potential. Systematic structure-function studies whereby the same P-MOF platform in multiple variables of substituents, metal, coordination mode and geometry will be critical for this purpose. Therefore, more quantified and standardized ways of measuring original structure and subsequent transformation of active site should be implemented. This have raised the requirement for in situ/ex situ characterization techniques as well as data processing abilities. Luckily, MOF based heterogeneous catalysis is an active, fast-evolving field, where new findings and inventions will help us further unveil their potential. Just like the success of phosphine ligands in homogeneous catalysis, an exciting future is anticipated for P-MOFs, where any progress on the above obstacles will be the new growth point of whole MOF catalysis field.
Acknowledgments
This work was supported by NPRP award (13S-0213-200353) from the Qatar National Research Fund and Youth Fund of the National Natural Science Foundation of China (No. 22301314). H.-C. Zhou acknowledges fnancial support from the Robert A. Welch Foundation through a Welch Endowed Chair (A-0030). S. T. Madrahimov also acknowledges Texas A&M University Qatar Responsive Research Seed Grants.
Conflict of interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Biographical Information
Wenmiao Chen finished Ph.D. studies in 2022 under the supervision of Prof. Hong-Cai Zhou, Texas A&M University (USA). Then he worked as a postdoctoral associate at Texas A&M University Qatar with Prof. Sherzod Madrahimov. Currently he is an associate professor in China University of Petroleum (East China). His research interests focus on the synthesis and catalytic application of MOFs.

Biographical Information
Peiyu Cai received his B.S. from Nanjing University in 2017. He finished Ph.D. studies in 2023 under the supervision of Prof. Hong-Cai Zhou, Texas A&M University (USA). He is currently a postdoctoral associate at TAMU. His current research interests focus on the synthesis and separation application of metal-organic frameworks.

Biographical Information
Hong-Cai “Joe” Zhou obtained his Ph.D. from Texas A&M University with Prof. F. A. Cotton. After a postdoctoral stint at Harvard University with R. H. Holm, he joined the faculty of Miami University, Oxford in 2002. He moved to Texas A&M University in 2008 and is currently the Robert A. Welch Chair in Chemistry. In June 2013, he became an associate editor for Inorganic Chemistry. His research focuses on the preparation and application of MOFs.

Biographical Information
Sherzod Madrahimov is currently an Associate Professor of Arts and Science Division at Texas A&M University at Qatar. He received his Ph.D. with Prof. John Hartwig at the University of Illinois Urbana Champaign. He then performed postdoctoral studies at Northwestern University with Prof. Son Binh T. Nguyen. His research focuses on immobilized single molecular catalysts to bridge the gap between homogeneous and heterogeneous catalysis.


