

Lithium Promotes Acetylide Formation on MgO During Methane Coupling Under Non-Oxidative Conditions
Graphical Abstract
Li doping of MgO critically improves selectivity toward C2-products in non-oxidative coupling of methane. The presence of Li favors the formation of magnesium acetylide and therefore is likely involved in C2 formation.

Abstract
A prototypical material for the oxidative coupling of methane (OCM) is Li/MgO, for which Li is known to be essential as a dopant to obtain high C2 selectivities. Herein, Li/MgO is demonstrated to be an effective catalyst for non-oxidative coupling of methane (NOCM). Moreover, the presence of Li is shown to favor the formation of magnesium acetylide (MgC2), while pure MgO promotes coke formation as evidenced by solid-state 13C NMR, thus indicating that Li promotes C−C bond formation. Metadynamic simulations of the carbon mobility in MgC2 and Li2C2 at the density functional theory (DFT) level show that carbon easily diffuses as a C2 unit at 1000 °C. These insights suggest that the enhanced C2 selectivity for Li-doped MgO is related to the formation of Li and Mg acetylides.
Introduction
Direct valorization of methane into higher hydrocarbons via methane coupling has been a longstanding research effort with high economic interest.1 Both non-oxidative (NOCM) and oxidative (OCM) methane coupling processes have been studied. Overoxidation of methane to COx species is the major route to byproducts in OCM, whereas NOCM suffers from coking.2 While each of these processes has specific challenges, they aim to generate similar products, i.e. C2 or aromatics, and are typically thought to involve different intermediates and reaction mechanisms.2-6 While surface sites are thought to be important to activate the C−H bond of methane, radicals and gas phase reactions are typically invoked for both OCM and NOCM.
 (1)
(1)Furthermore, the catalytic function of MgO is presumed to be connected to defect sites rather than the MgO phase itself.7 While the involvement of [Li+O−⋅]MgO is one prevalent rationale for catalytic activity, the nature of the active sites is still a matter of debate,3, 8, 11 considering that materials containing neither Li or Mg are also active and selective in OCM toward C2 products.3 A possible alternative mechanism involves the heterolytic C−H activation of methane on a [Mg2+O2−] site, yielding a surface OH group and a Grignard-type Mg-methylate ([HO−(Mg−CH3+)]) intermediate. Further reaction with O2 is proposed to liberate the free methyl radicals, which then couple in the gas phase to generate carbon-carbon bonds.11
In NOCM and related processes, which operate at higher temperatures, metal carbides are formed under reaction conditions and can also be employed as catalytic materials.12-14 In fact, recent work from our group has shown that bulk carbidic carbons in Mo and W carbides can readily exchange with methane present in the gas phase, due to a fast carbon diffusion at these reaction conditions. This study also suggests that carbon homologation may not necessarily involve gas phase processes and that coupling of hydrocarbyl ligands can also take place at the surface of the metal carbide materials.12
The involvement of specific mechanisms for OCM and NOCM is certainly consistent with the respective operating conditions, in particular the presence or absence of molecular oxygen as well as the different operating reactor temperatures (700–1100 °C). While thermodynamic limitations in NOCM require the use of high reaction temperatures up to 1100 °C,2 OCM is typically operated at 800 °C. Note however that OCM is so exothermic that increase of temperature in the catalyst bed up to 1100 °C has been reported.15
Bearing the similarities in product and operation temperature in mind, we decided to examine the performance of Li/MgO and MgO under NOCM conditions. In particular, this work shows that, under NOCM conditions, Li/MgO is particularly efficient to promote carbon-carbon bond formation and generate acetylide intermediates. The observation of these intermediates correlates with the high C2 selectivity, suggesting a relation between them.
Results and Discussion
First, Li/MgO containing 6 wt % Li was synthesized as previously reported.16 Next, Li/MgO was contacted with a flow of methane at 1000 °C (40 sccm feed gas, 200 mg cat., 10 % CH4/Ar). Under these conditions, ethane was observed as major product with initial C2 selectivities reaching 30 % (Figure S2). The C2 selectivity declined over the course of 2 h and stabilized at around 10 % C2 selectivity with a steady-state CH4 conversion of ca. 5 %, showing that Li/MgO can indeed participate in carbon-carbon bond formation under non-oxidative conditions without the need of molecular oxygen. The residual selectivity is ascribed to coke formation.
Isotope labelling experiments were then conducted in batch with the goal to track the state of the spent catalyst (Figure 1a). 200 mg of Li/MgO (6 wt % Li) or MgO were loaded into a quartz reactor which was then filled at room temperature with 200 mbar of 13CH4 (molar ratio 1 13CH4 : 2 Li : 6 MgO for Li/MgO and 1 13CH4 : 6 MgO for MgO) and heated to 1000 °C for 2 h with aliquots drawn after 1 and 2 h. The reactor was evacuated and refilled at room temperature after 2 h due to significant methane consumption and was allowed to react at 1000 °C for another 0.5 h.

Methane coupling with Li/MgO under non-oxidative conditions at 1000 °C. (a) Reaction Setup: the batch reactor is filled with the corresponding catalyst and 200 mbar of 13CH4 before heating to 1000 °C and aliquots are drawn after 1 h, 2 h and 2.5 h. GC traces are shown for Li/MgO (b) and MgO (c). The right side (after dashed blue line) corresponds to the trace of m/z=27 to allow for better signal intensity.
Under these conditions, a rapid consumption of methane can be observed (full conversion within 2 h), while ethane, ethylene and acetylene are detected in the gas phase (Figure 1b). The highest selectivities are observed for ethylene and acetylene while ethane selectivity is comparatively low. Surprisingly, the spent Li/MgO yielded a white powder without visible evidence for coking. Notably, the quartz glass reactor showed signs of devitrification, which is not observed in the absence of Li indicative of intercalation of “Li2O” in the silica-glass wall and thereby loosing Li from the catalyst.17 Considering the thermodynamic equilibrium among C2 species at 1000 °C (see Figure S1), the observed C2 distribution is in line with expectations. Besides C2 species, CO2 and H2O are detected, showing that the oxygen of the oxide support is accessible and can participate in the reaction.
The corresponding reaction with MgO was further investigated (Figure 1c), where both CH4 consumption and C2 product selectivities are significantly lower. This observation parallels what has been observed in OCM where Li doping is crucial for high C2 selectivities.18, 19 Contrary to Li/MgO, the spent MgO turns black, visibly showing coking while no devitrification of the quartz glass reactor was observed.
To understand the difference in catalytic activity and appearance of these two solids post-reaction, they were analyzed by solid-state 13C magic-angle-spinning (MAS) NMR spectroscopy. The 13C MAS-echo NMR spectrum of post-reaction MgO (Figure 2a) is dominated by an intense 13C signal at 129 ppm, consistent with the formation of graphitic carbonaceous deposits,20 the color change of the material post-reaction and the poor C2 selectivity. In contrast, the 13C MAS echo NMR spectrum of Li/MgO (Figure 2b) shows an intense narrow 13C signal at 170 ppm along with additional signals at 14 and 3 ppm. A 13C{1H} cross-polarization (CP)-MAS spectrum (Figure 2c), acquired at low temperature (100 K) in order to improve signal sensitivity and to mitigate the influences of surface dynamics, shows an additional weak signal at 129 ppm as well as a distribution of signals in the 14–34 ppm region. The low relative intensity of the 13C signal at 129 ppm from coke or related aromatic species in spent Li/MgO compared to MgO indicates that Li effectively suppresses most coke formation and contributes to the enhanced C2 selectivity of the Li/MgO material. The 13C NMR signal at 170 ppm is attributed to MgC2 based on literature reports21 and the formation of the MgC2 species is only observed in the catalyst material containing Li (see below). Close inspection of the 13C{1H} CPMAS and single-pulse 13C spectra of the Li/MgO acquired under the same conditions (Figure S5) show that there are in fact two signals that overlap in this region, with very similar chemical shifts (170 vs 170.5 ppm) but different linewidths (3 vs 5 ppm) that are tentatively attributed to bulk vs near-surface Mg acetylide species, respectively. The minor 13C signals at 3 and 14 ppm could arise from Mg alkyl species, which tend to exhibit shielded 13C chemical shifts.22 The signal at 34 ppm is noteworthy and suggests the formation of alkyl chains that are at least three carbons long, which could be associated with alkyl magnesium or alkylated aromatic surface species. The 1H MAS NMR spectrum of spent Li/MgO (Figure S3) shows a range of relatively narrow 1H signals from 0.4 to 3.2 ppm, indicating the presence of a distribution of alkyl species, as well as broad signals at 5.2 and 7.4 ppm, consistent with the presence of surface-bound water and aromatic species, respectively.

Solid-state 13C MAS-echo NMR spectra of spent a MgO and b Li/MgO after reaction in batch for 2.5 h at 1000 °C. Spectra were measured at 9.4 T, 298 K, and 10 kHz MAS. c Solid-state 13C{1H} CPMAS NMR spectrum of spent Li/MgO acquired at 14.1 T, 100 K, 12 kHz MAS, and a 13C−1H contact time of 2 ms. Asterisks indicate spinning sidebands. d Solid-state single-pulse 6Li MAS NMR spectra of spent (red) and pristine (black) Li/MgO catalyst acquired at 16.4 T, 298 K, and 16 kHz MAS.
While no lithium acetylide species, expected at around 195 ppm,23 was observed in the 13C MAS NMR spectra of the spent catalyst, probably due to low sensitivity and concentration, solid-state 6Li MAS NMR analyses of pristine and spent Li/MgO reveal the presence of Li−C species as indicated by the doublet feature likely due to the coupling of 6Li and 13C nuclei (Figure 2d). A prominent 6Li signal at 0.0 ppm observed both before and after reaction corresponds to LiI bound in a mixed metal oxide LixMgyOz.
After reaction, additional 6Li NMR signals are detected at 0.8 and 1.4 ppm, which can be attributed to distinct types of Li acetylide or related organolithium species.24, 25 The solid-state 6Li and 13C results overall show that Li and Mg acetylides are formed in situ and are stable post-reaction under inert conditions. This finding indicates a close relation between in situ MgC2 formation and C2 selectivity, hence providing a potential rationale for enhanced C2 selectivity of the Li-doped MgO as the formation of MgC2 is not observed in the absence of Li.
 (2)
(2)Similarly to calcium, the reaction of Mg acetylides and water will yield acetylene (Eq. 2).26 Under NOCM, this reaction could play a role in product formation to some extent as the formation of water has been observed under our reaction conditions.
 (3)
(3) (4)
(4)Li2C2 is known to possess an exceptional stability (ΔfH°(Li2C2)=−14.1 kcal mol−1) compared to other alkali and earth alkali acetylides (ΔfH°(MgC2)=+19.0 kcal mol−1) and its formation was reported as early as 1896.28 To be more realistic with regard to the experimental conditions, standard pressure for methane and 10−2 bar pressure for H2O and H2 were considered (see Figure S4 for alternative plots under standard conditions). As expected, the formation of Li2C2 is much more favorable than MgC2 (Figure 3) and becomes favorable around 900 °C while MgC2 is only expected to be a metastable phase at reaction temperature. Based on catalyst bed temperatures in OCM of up to 1100 °C, it is proposed that the formation of Li2C2 induces the formation of thermodynamically disfavored MgC2. The role of Li is thus ascribed to inducing formation of MgC2.

Gibbs free energy of formation of Li2C2 and MgC2 from the reaction of methane with the corresponding oxides as a function of temperature.
Considering that Li and Mg acetylides can be considered as a subclass of metal carbides and that both have been shown to be formed in situ, they might play a similar role in methane coupling reactions. These findings parallels the behavior observed for Mo2C and WC, where bulk carbon can participate in C2 product formation and diffusion coefficients of carbidic carbon are proposed descriptors for carbidic carbon exchange dynamics, and can describe the extent of exchange between methane in the gas phase and metal carbide (surface and bulk), resulting in incorporation of carbidic carbon in the products.12 In the case of alkaline (earth) metal acetylides, the bulk structures feature embedded C2 units (C−C bond length of 1.2 Å), thus the direct evolution of C2Hx (x=2,4,6) products by hydrogenation or interaction with adsorbed H is probable. To gain insight into the influence of the presence of acetylides on the catalytic performances of Li/MgO, the diffusion mechanism of carbon in MgC2 and Li2C2 was explored employing metadynamics at the density functional theory (DFT) level (see Supporting Information Section 5 for computational details).29-35 The diffusion of a carbon atom at 1000 °C through an approximately 10×10×10 Å cell of metal acetylide was considered. A history-dependent bias was applied along two collective variables (CV): i) the distance of a selected carbon atom to the geometric center of the cell (d), and ii) the coordination number of this carbon atom to the other carbon atoms CN(C−C). Applying this set of CVs, carbon diffusion pathways were explored while enabling C−C bond cleavage.
In MgC2, carbon diffusion takes place in the plane of the C2 units (Figure 4a, initial). The biased carbon (marked in red) diffuses as a C2 unit in the bulk structure, which is energetically preferred over C−C bond cleavage. A transient C4 chain is formed between the diffusing C2 and the neighboring C2 unit (Figure 4a, C4). The average C−C bond lengths range from 1.26 Å to 1.33 Å upon C4 chain formation. This transient C4 chain then breaks and a substitutional diffusion36 of the C2 fragment takes place to reach a final configuration identical to the initial one (Figure 4a, final). The overall free energy barrier for this diffusion process in MgC2 is 54.0 kcal mol−1, which is associated to a carbon diffusion coefficient  =1.7×10−15 m2 s−1, evidencing that carbon diffusion in MgC2 is accessible at 1000 °C (Figure 4b).
=1.7×10−15 m2 s−1, evidencing that carbon diffusion in MgC2 is accessible at 1000 °C (Figure 4b).

Snapshots of the metadynamics simulations of carbon diffusion in MgC2 a and Li2C2 c corresponding to initial, intermediate and final states. Mg is illustrated in pink and carbon in grey. The diffusing C2 units are highlighted in dark red. Reconstructed free energy surfaces along distance d and CN(C−C) collective variables for MgC2 b and Li2C2 d and associated free-energy barriers (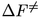 ) and diffusion coefficients (
) and diffusion coefficients ( ).
).
At the same temperature, the Li2C2 is in molten phase as expected from the bulk melting temperature (Tmp=452 °C),37 which leads to a facile diffusion of the C2 units (Figure 4c) featuring an average length of 1.27 Å of the C−C bond. The resulting free energy barrier for C2 diffusion in Li2C2 is significantly lower at 12.6 kcal mol−1, and the associated diffusion coefficient ( =1.1×10−8 m2 s−1) reflects the fast diffusion of carbon in Li2C2 at 1000 °C (Figure 4d).
=1.1×10−8 m2 s−1) reflects the fast diffusion of carbon in Li2C2 at 1000 °C (Figure 4d).
In both materials, C2 units diffuse without undergoing C−C bond cleavage. The ease of C2 diffusion suggests facilitated exchange of carbidic carbon with the gas phase for both acetylides, though with much faster diffusion for Li2C2 compared to MgC2. The persistence of the C2 moiety in the bulk is reflected on the reactivity of the acetylide material leading to increased C2 selectivity.
Conclusion
This study shows that the archetypical OCM catalyst Li/MgO can efficiently promote carbon-carbon bond formation under NOCM conditions yielding C2 products, without significant coke formation. Analysis of Li/MgO post-reaction clearly evidences the formation of MgC2 in the presence of Li, while pure MgO favors coke formation (as do many other oxides). It is proposed that the presence of Li promotes the formation of MgC2, probably through the formation of Li2C2, which explains the origin of the enhanced C2 selectivity of Li/MgO as opposed to pure MgO.38, 39 This increase in selectivity can be partially rationalized by the increased carbon mobility where the carbon of the acetylide diffuses as C2 unit as shown by metadynamics simulations, though the mechanism of carbon-carbon bond formation remains to be explored. While MgC2 has thus far not been reported as a catalyst under OCM conditions, one may propose that MgC2 species are formed as transient intermediates contributing to the higher observed C2 selectivity in the presence of Li-doping. The formation of organo-lithium/magnesium species on Li/MgO has been previously proposed11 and could be an important step toward carbon-carbon bond formation in the bulk, leading to C2 (acetylide) unit formation. Notably, this process is reminiscent of the acetylene production based on calcium carbide, which has been a multi-ton scale process since the 70s and remains the most important production route to the present day.40, 41
We think that the discussed parallels between OCM and NOCM open alternative perspectives for the development of catalytic materials. The involvement of metal carbides or acetylides seems to be a recurring theme in methane coupling where carbidic carbon exchange dynamics has to be considered. We are currently pursuing these lines of research in our pursuit to develop alternative catalytic systems.
Acknowledgments
We thank Shell Global Solution International B.V. for financial support. Z. J. B. acknowledges support from an ETH Career Seed Award as well as ETH+ Project SynMatLab. P.-A. Payard and Q. Pessemesse are grateful to Université Lyon 1 and the Region Auvergne Rhone Alpes for financial support. P.-A. P. and Q. P. are grateful to the CCIR of ICBMS, PSMN, GENCI-TGCC (Grant A0120813435) for providing computational resources and technical support. Open Access funding provided by Eidgenössische Technische Hochschule Zürich.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.

