

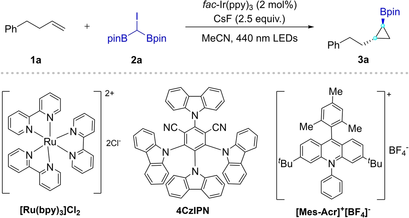
Photocatalyzed Borylcyclopropanation of Alkenes with a (Diborylmethyl)iodide Reagent
Graphical Abstract
A stable, multifunctional (diboronmethyl)iodide reagent (CH(Bpin)2I) was synthesized for the photoinduced cyclopropanation of alkenes, providing an array of 1,2-substituted cyclopropylboronates. The transformation exhibits operational simplicity, broad substrate scope, excellent functional-group compatibility, high chemo- and diastereoselectivity, and practicality for the late-stage modification of complex molecules.

Abstract
Cyclopropane skeletons play a prominent role in the development of organic synthesis and pharmaceutical chemistry. Herein, we report the design and synthesis of a stable, multifunctional (diborylmethyl)iodide reagent (CHI(Bpin)2) for the photoinduced cyclopropanation of alkenes, providing an array of 1,2-substituted cyclopropylboronates in good yields. This α-haloboronic ester can be readily synthesized on a multigram scale from commercially available starting materials. Furthermore, the protocol displays high chemo- and diastereoselectivity, excellent functional-group tolerance, and allows for late-stage borylcyclopropanation of complex molecules. Mechanistic studies reveal that the borylcyclopropanation proceeds through a radical addition/polar cyclization pathway mediated by the photocatalyst fac-Ir(ppy)3 and visible light.
Introduction
Cyclopropanes represent important structural motifs found in pharmaceuticals and biologically active molecules, showing unique properties, such as increasing metabolic stability, enhancing potency, and decreasing plasma clearance.1 Additionally, they are highly versatile building blocks for a wide range of bond-forming reactions in organic synthesis.2 Given their significance, considerable effort has been devoted to developing efficient methods for the preparation of such cyclopropyl derivatives over the past decade.2a, 3, 4, 5 Among various synthetic strategies, direct [2+1]-type cyclopropanation of alkenes is the most attractive and efficient approach to obtain these versatile molecules (Scheme 1a).6 In this context, C1 reagents have been developed for direct construction of cyclopropyl-based building blocks. Classically, dihalomethanes7 and diazo compounds8 have been extensively explored for the cyclopropanation of olefins, by processes involving the Simmons-Smith reaction pathway, carbenoid intermediates, or radical carbenoid species. Although these reactions are well-established, they have several shortcomings. 1) These dihalide or diazo carbenoid precursors are not easily prepared, and they are unstable and difficult to store for long periods of time because of their high reactivity. 2) These methods are often lacking in operational simplicity or broad functional group compatibility, hindering their potential for industrial development. 3) They are usually not suitable for late-stage modification of complex molecules because these reactions require large amounts of highly reactive chemicals and multiple additives. 4) These approaches are limited by a lack of effective control over chemo- and diastereoselectivity. To solve the aforementioned problems, new synthetic strategies have been developed for the efficient synthesis of cyclopropanes. In 2013, Hu and co-workers developed a metal-free cyclopropanation of alkenes for the synthesis of gem-difluorocyclopropanes using (bromodifluoromethyl)trimethylsilane (TMSCF2Br) as a difluorocarbene source.9 Subsequently, Rovis's group reported the use of N-enoxyphthalimides as a unique one-carbon source for the cyclopropanation of activated alkenes.10 In 2018, the Molander group disclosed a photocatalytic cyclopropanation process using a triethylammonium bis(catecholato)iodomethylsilicate as a C1 reagent.11 In addition, Wu, Radius, and Marder described the copper-catalyzed borylcyclopropanation of alkenes with B2pin2 using CO as the C1 component, affording cyclopropyl bis(boronates).12 In 2022, Yamanaka and Tobisu used acylsilanes as electron-rich carbenes for the Pd-catalyzed siloxycyclopropanation of alkenes.13 In the same year, Wang synthesized cyclopropanols or cyclopropylamines with high diastereoselectivity by a Ti-catalyzed cyclopropanation of alkenes with carboxylic acid derivatives.14 Borylcyclopropanes are important synthetic intermediates for the preparation of functionalized cyclopropanes because their carbon-boron bonds can be efficiently converted to carbon-carbon and other carbon-heteroatom bonds.15 Previous synthetic methods mainly focus on the borylcyclopropanation of alkenes with diiodomethylpinacol boronate16 or diazo compounds,17 and borylative cyclization of allylic compounds with B2pin2 catalyzed by transition metals.18 While these are significant advances, only recently has Charette reported a photocatalyzed single-electron transfer approach for the borylcyclopropanation of styrenes with CHI2Bpin, providing corresponding borocyclopropanes with moderate diastereoselectivity.19 Therefore, the development of new synthetic techniques or novel C1 reagents for the synthesis of stereodefined borylcyclopropanes is valuable.

Direct cyclopropanation reactions.
gem-Diborylalkanes have received much recent interest as versatile building blocks in organic synthesis because they can be used in multi-step functional transformations or cross-coupling reactions, providing a practical synthesis platform for the construction of complex molecules.20 These bifunctional units have been widely used in new synthetic strategies and reaction modes.21 In general, there are mainly two modes of reactivity in these transformations. One category is monodeborylative cross-coupling of gem-diborylalkanes through the generation of α-borylalkylmetal species or an α-boryl carbanion. [22] Another category is the deprotonation of diborylalkanes to afford gem-diboryl carbanion species which can further cross-couple with various organo-electrophiles.23 However, a photocatalytic approach for the synthesis of cyclopropanes using gem-diborylalkanes as C1 reagents has not been explored. We present herein the photoinduced borylcyclopropanation of alkenes with a (diborylmethyl)iodide reagent,24 which can be readily prepared from bis[(pinacolato)boryl]methane in a single step (Scheme 1b), noting that bis[(pinacolato)boryl]methane can be prepared directly by Cu-catalyzed borylation of CH2Cl2.25 Our visible-light-induced strategy provides access to a series of stereodefined cyclopropyl boronic esters in good yields under mild conditions.
Results and Discussion
We set out to investigate the possibility for a photocatalyzed borylcyclopropanation using 4-phenyl-1-butene (1 a) and 2,2′-(iodomethylene)bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane) (2 a) as model substrates (Table 1). Systematic optimization experiments demonstrated that cyclopropyl boronic ester 3 a, as a single isolated regioisomer which was determined by NMR spectroscopy, was obtained in 78 % yield using 2 mol% of fac-Ir(ppy)3 as the photocatalyst (PC), 2.5 equiv. of CsF as the base in MeCN under irradiation with a 440 nm blue LED at room temperature for 24 h (Table 1, entry 1). The use of KF as the base showed much lower reactivity for this transformation (Table 1, entry 2). Other bases, such as Cs2CO3, K3PO4, TBAF, KOAc, NaOtBu, and Et3N, failed to give the desired borylcyclopropanes (Table 1, entries 3–8). With [Ru(bpy)3]Cl2, 4CzIPN, or Eosin Y in place of fac-Ir(ppy)3, 3 a was formed in a reduced yield (Table 1, entries 9–11). In addition, when [Mes-Acr]+[BF4]− was used as a PC, the corresponding products were not detected by GC-MS (Table 1, entry 12). While 3 a was formed in 64 % yield with dichloromethane (DCM) as the solvent (Table 1, entry 13), other commonly used solvents, such as THF and DMF, were ineffective for this photoinduced reaction (Table 1, entries 14 and 15). Finally, control reactions revealed that the PC, additive, and visible light are all essential for the borylcyclopropanation (Table 1, entries 16–18).
|
||
Entry |
Variation from standard conditions |
Yield of 3 a [%][b] |
|---|---|---|
1 |
None |
89(78)[c] |
2 |
KF instead of CsF |
37 |
3 |
Cs2CO3 instead of CsF |
0 |
4 |
K3PO4 instead of CsF |
0 |
5 |
TBAF instead of CsF |
0 |
6 |
KOAc instead of CsF |
0 |
7 |
NaOtBu instead of CsF |
0 |
8 |
Et3N instead of CsF |
0 |
9 |
using [Ru(bpy)3]Cl2 as PC (2 mol%) |
79 |
10 |
using 4CzIPN as PC (5 mol%) |
52 |
11 |
using Eosin Y as PC (5 mol%) |
68 |
12 |
using [Mes-Acr]+ [BF4]− as PC (5 mol%) |
0 |
13 |
using DCM as solvent |
64 |
14 |
using THF as solvent |
0 |
15 |
using DMF as solvent |
0 |
16 |
without CsF |
0 |
17 |
without PC |
0 |
18 |
in the dark |
0 |
- [a] Reaction conditions, unless otherwise stated: 4-phenyl-1-butene 1 a (0.30 mmol, 1.0 equiv.), CHI(Bpin)2 2 a (0.36 mmol, 1.2 equiv.), photocatalyst (2–5 mol%), base (0.75 mmol, 2.5 equiv.), solvent (1.0 mL), 24 h, 440 nm blue LED, 25–40 °C, under argon. [b] The yields of 3 a were determined from the crude reaction mixtures by GC-MS analysis vs. a calibrated internal standard and are averages of two runs. [c] Yield of 3 a after chromatographic workup.
Under the optimal reaction conditions, we proceeded to investigate the substrate scope of this borylcyclopropanation reaction using various alkenes. As shown in Scheme 2, a series of alkene substrates with various carbon chains, including isopropyl, tert-butyl, cyclohexyl, and cyclopentyl, were all successfully converted to the corresponding cyclopropanyl boronate esters in 67–82 % isolated yields (3 b–3 g). Alkenes bearing trifluoromethoxy (1 h), thiophenyl (1 i), or methoxy (1 j) groups could be used under our conditions to provide the desired products in useful yields (3 h–3 j). Halide substituents (F, Cl, Br and I) on the benzene rings or alkyl chains of the substrates were compatible with the borylcyclopropanation reaction (3 k–3 p), and these cyclopropanes with halogen functional groups have potential applications in subsequent cross-coupling transformations. A diverse set of ester-based substrates (1 q–1 t) were also appropriate reaction partners. Functional groups, including amide (1 u), phthalimide (1 v), dimethylaminphenyl (1 w), or benzenesulfonamide (1 x), were all well tolerated in this transformation, affording the corresponding cyclopropyl boronic esters in 61–73 % yields. The configuration of 3 v was further confirmed by single-crystal X-ray diffraction.26 An alkene bearing a silyl-protected alcohol reacted with 2 a, yielding the product 3 y in 72 % yield. Notably, when an alkene bearing an OTs group was subjected to our conditions, we did not detect the corresponding products by NMR or GC-MS, but iodine-substituted borylcyclopropane (3 z) was formed instead. The borylcyclopropanation of a vinylsilane and a vinylboronate with 2 a gave 1,2-difunctionalizated products 3 aa and 3 ab in 77 % and 84 % yields, respectively. Interestingly, alcohols and ketones have no effect on the reactivity, demonstrating the excellent functional group tolerance of this reaction. Using a non-conjugated diene as the substrate, selective borylcyclopropanation occurred at the terminal double bond, affording 2-(2-(cyclohex-3-en-1-ylmethyl)cyclopropyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 3 ae in 74 % yield. Interestingly, reaction with norbornene 1 af afforded 3 af in 41 % yield. Substrates with naphthyl or heterocyclic cores, such as thiophene, benzofuran, indole, and furan, were also compatible with our standard conditions, affording the borylated cyclopropanes in moderate to good yields (3 ag-3 al). A 5.0 mmol scale reaction with 1 a and 2 a afforded 3 a in 68 % isolated yield (see Supporting Information). Unfortunately, the Me-substituted analogue of 2 a, namely 2,2′-(1-iodoethane-1,1-diyl)bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane) 2 b (CH3CI(Bpin)2), did not yield the expected cyclopropanation product; instead, CH2=C(Bpin)2 and gem-diborylalkane MeCH(Bpin)2 were detected by GC-MS.

Borylcyclopropanation of alkenes. Reaction conditions: a mixture of alkene 1 (0.3 mmol, 1.0 equiv.), 2 a (1.2 equiv.), fac-Ir(ppy)3 (2 mol%), and CsF (2.5 equiv.) in MeCN (1 mL) was stirred for 24 h, irradiating with a 440 nm blue LED at 25–40 °C under argon; isolated yield after chromatography.
As cyclopropanes are ubiquitous in bioactive molecules and pharmaceuticals, we anticipated that this borylcyclopropanation could provide a general method for the rapid construction of three-membered rings for medicinally relevant products. Therefore, we carried out a number of functionalizations of complex molecules (Scheme 3). L–Menthol derivative 1 ar was subjected to our reaction conditions, delivering 3 ar in 72 % yield. The borylcyclopropanation was successful for ketoprofen, diacetone-D-glucose, and epiandrosterone under the standard conditions, delivering 3 as, 3 at, and 3 au in 63, 51, and 58 % yields, respectively. Compound 1 av, derived from β-estradiol, exhibits good reactivity for the catalyzed photochemical borylcyclopropanation. Oxaprozin reacts to form the borylcyclopropanated product 3 aw in 70 % yield. In addition, several natural product derivatives, including pregnenolone 1 ax, vitamin E 1 ay, and diosgenin 1 az, underwent photoinduced borylcyclopropanation to afford the corresponding products in 48 %-67 % yield. This synthetic strategy will provide a new and practical platform for the discovery of new drugs containing cyclopropane moieties.

Applications in the functionalization of complex molecules. Reaction conditions: a mixture of alkene 1 (0.3 mmol, 1.0 equiv.), 2 a (1.2 equiv.), fac-Ir(ppy)3 (2 mol%), and CsF (2.5 equiv.) in MeCN (1 mL) was stirred for 24 h, irradiating with a 440 nm blue LED, 25–40 °C, under argon; isolated yield after chromatography.
Next, we focused our attention on exploring the mechanism of the reaction. First, without addition of CsF, only a trace amount of hydro-deiodinated product 5 a was detected; all of the starting materials 1 a and 2 a were recovered, and possible byproduct 4 a, which could be generated by atom transfer radical addition, was not observed in this process (Scheme 4a). Thus, CsF plays a crucial role in the reaction. Additionally, 11B NMR spectroscopic studies in CD3CN of the reaction of 2 a with CsF (see Supporting Information),27 show a broad peak at 32.23 ppm, characteristic of a C-Bpin species, a very weak and broad peak at ca. 22 ppm, typical of an O-Bpin species, a broad doublet at 6.15 ppm (J=68 Hz) likely indicative of a 4-coordinate boron compound, and a sharp triplet at 5.01 ppm (J=22 Hz) clearly indicative of a fairly symmetric 4-coordinate boron compound. Although ESI-HRMS studies show a peak at (m/z)=335.0095, which is clearly consistent with the formula C7H13B2O2F3I (calc'd m/z=335.0098) appropriate for the [CHI(Bpin)(BF3)] anion 2 a′, in which it is also possible that one fluoride moiety weakly bridges between BF2 and Bpin groups, as depicted in Scheme 4d, the 11B NMR spectrum is consistent with the formation of several additional species including [F2Bpin]− and an adduct of FBpin (perhaps with MeCN), which could arise from cleavage of one C−B bond, resulting in the formation of the [CHI(Bpin)]− anion (see below). The small amount of O-Bpin species may well be B2pin3 (i.e., pinB-OCMe2CMe2O-Bpin), formed from disproportionation of FBpin.27c We performed the borylcyclopropanation of 1 a with “isolated” 2 a′ (this likely being a mixture of species, as noted above) using fac-Ir(ppy)3 as the PC in MeCN, providing the product 3 a in 81 % yield. Thus, species formed from reaction of CsF with 2 a are clearly involved in the reaction.

Mechanistic studies.
The radical character of the borylcyclopropanation reaction was confirmed by a series of radical trapping experiments (Scheme 4b), again using “isolated” 2 a′. Notably, two radical adducts 6 a and 7 a were detected by high resolution mass spectrometry. These results imply the presence of a carbon-centered radical containing I and Bpin in this process.28 Subsequently, we conducted Stern–Volmer quenching experiments. As shown in Scheme 4c, the luminescence of fac-Ir(ppy)3 at λmax=526 nm is clearly quenched by 2 a in the presence of CsF, whereas in the absence of CsF, the quenching is inefficient. These studies again indicate that 2 a′, or a related species formed from reaction of 2 a with CsF (see above), is likely the species responsible for quenching of luminescence.
With these preliminary data in hand, we propose a plausible mechanism for the reaction (Scheme 4d). Initially, 2 a reacts with CsF to form a mixture of species including the trifluoroborate complex 2 a′. This species may also lead to the formation of the [CHI(Bpin)]− anion. Subsequently, the carbon-centered radical CHI(Bpin) I thus formed and an IrII complex are generated by reductive quenching of the excited iridium photocatalyst by either trifluoroborate complex 2 a′ or the [CHI(Bpin)]− anion 2 a’′. The alkyl radical (I) adds to the alkene giving II, which undergoes SET from the reduced iridium catalyst to give carbanion III and the ground state IrIII photocatalyst. Finally, the borylcyclopropane product 3 is formed through a polar 3-exo cyclization of the resulting carbanion III, and iodide ion is generated.
The fact that reaction of 2 a with CsF generates a mixture of species, and that we have not succeeded in isolating 2 a′ as a pure material, preclude our ability to obtain reliable redox potentials for the actual active species. Thus, we emphasize the speculative nature of our proposed mechanism and that other mechanisms including oxidative quenching as a competing activation mode cannot be completely ruled out at this early stage. However, we note similarities with the mechanism proposed by Molander et al.11 for a photocatalyzed cyclopropanation using an anionic bis(catecholato)iodomethyl silicate as the C1 reagent. While these authors focused on 4CzIPN as the photocatalyst, they also showed that Ir(ppy)3 was effective, giving their product in 74 % yield.29 Likewise, in our process, both Ir(ppy)3 and 4CzIPN are effective photocatalysts (Table 1).
Conclusion
The (diborylmethyl)iodide reagent CHI(Bpin)2, readily available from diborylmethane, itself readily available from CH2Cl2, has been used effectively in the selective borylcyclopropanation of alkenes under photoredox catalysis. This novel strategy leads to a variety of cyclopropylboronates in good yields and with excellent diastereoselectivities, and is tolerant to a wide range of functional groups. Preliminary mechanistic studies suggest that the reaction proceeds through radical addition/polar cyclization between an α-iodoboryl carbon-centered radical and the alkene. We anticipate that this approach will simplify the synthesis of borylcyclopropanes for research in pharmaceutical chemistry and organic synthesis.
Acknowledgments
T.B.M. thanks the Julius-Maximilians-Universität Würzburg for financial support. Z.W. thanks the China Scholarship Council (CSC) for a Ph.D. Scholarship. J.H. thanks the Alexander von Humboldt Foundation for a Postdoctoral Fellowship and the National Natural Science Foundation of China (21901114) for financial support. We thank AllyChem Co. Ltd. for a generous gift of diboron(4) reagents. Open Access funding enabled and organized by Projekt DEAL.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.

