

Production of Hydroxy Acids: Selective Double Oxidation of Diols by Flavoprotein Alcohol Oxidase
Graphical Abstract
Triple action: The flavoprotein alcohol oxidase (AOX*) facilitates the double and triple oxidations of diols. Interestingly, depending on the diol substrate, these reactions result in formation of either lactones or hydroxy acids. Such biocatalytic route towards hydroxy acids has potential for the preparation of polyester building blocks.

Abstract
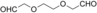
Flavoprotein oxidases can catalyze oxidations of alcohols and amines by merely using molecular oxygen as the oxidant, making this class of enzymes appealing for biocatalysis. The FAD-containing (FAD=flavin adenine dinucleotide) alcohol oxidase from P. chrysosporium facilitated double and triple oxidations for a range of aliphatic diols. Interestingly, depending on the diol substrate, these reactions result in formation of either lactones or hydroxy acids. For example, diethylene glycol could be selectively and fully converted into 2-(2-hydroxyethoxy)acetic acid. Such a facile cofactor-independent biocatalytic route towards hydroxy acids opens up new avenues for the preparation of polyester building blocks.
Oxygen-containing heterocycles (O-heterocycles) form a class of compounds proven to be relevant in the polymers, fuel, and medical fields.1-4 Some of these O-heterocycles, such as lactones, and in particular 1,4-dioxan-2-one can be used for synthesis of biodegradable polyesters that find countless clinical applications thanks to their biocompatibility and strength properties.5, 6 Bioabsorbable polymers derived from 1,4-oxathian-2-one possess similar characteristics to polydioxanone, but they are not as studied and commercially used, probably because of the poor yield in the synthesis of 1,4-oxathian-2-one.7 The chemical routes to synthetize some of these compounds often require expensive transition-metal catalysts (Au or Pd catalysts), which increase the production costs.8 Moreover biodegradable polymers for biomedical applications need to be metal free.9 Enzymatic synthesis of lactones as polyester building blocks gained great interest as an environmentally sustainable alternative to current chemical methods. The most common biocatalysts to produce lactones are Baeyer–Villiger monooxygenases (BVMOs) and alcohol dehydrogenases (ADHs).10-13 These two classes of enzymes require cofactor NAD(P)H regeneration, making these strategies less suitable for commercial applications. Oxidases are an attractive class of enzymes for the production of bulk chemicals since they use oxygen either as an oxidant without the need to regenerate cofactors.14 Alcohol oxidases are a subclass of oxidative enzymes containing either a copper or flavin adenine dinucleotide (FAD) as a prosthetic group that often has a broad substrate spectrum, including primary and secondary alcohols, aldehydes, and ketones.15 A remarkable example of the power of flavoprotein oxidases is the 5-hydroxymethylfurfural (HMF) oxidase discovered and engineered by our group. This FAD-containing biocatalyst can oxidize 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid in three consecutive oxidations.16, 17 Another recent demonstration of the biocatalytic potential of a flavoprotein alcohol oxidase was recently reported by Turner and co-workers. They showed that choline oxidase can be engineered towards a wider substrate acceptance, for the selective oxidation of primary alcohols to aldehydes.18
The FAD-containing alcohol oxidase from the white-rot basidiomycete Phanerochaete chrysosporium (PcAOX) was recently characterized and engineered by our group for improved activity towards glycerol.19 The rational engineering study resulted in the variant F101S, optimized for glycerol conversion. The aim of this work was to explore and expand the potential of this improved variant. Herein, the attention was directed to diols which represent an industrially important class of chemical compounds as they are relatively cheap and can be transformed into either lactones or hydroxy acids, which in turn are building blocks for biodegradable polymers. Except for a focus on diols as substrates, we also tested other alcohols (primary and secondary) and aminoalcohols. Such a large variety of different substrates was explored to probe the substrate acceptance profile of F101S-PcAOX, but also to understand how catalyzes the double oxidation of alcohols into carboxylic acids. The most striking discovery is the ability of F101S-PcAOX to selectively perform double oxidations on one of two hydroxy groups for a subset of diols.
The initial experiments were performed with a small set of diols, aimed to verify which variant of PcAOX (wild-type PcAOX or the F101S variant) was more active (see the Supporting information). NMR analysis proved to be the best method for analysis of these types of compounds. This approach is fast and it allowed a quantitative and qualitative analysis of the formed products. Table 1 presents the list of substrates tested and the product yields as determined by 1H NMR spectroscopy. The results revealed that the F101S mutant outperformed the wild-type enzyme, in line with its wider active site. F101S-PcAOX (referred to as AOX* in the rest of the manuscript) was selected for further investigation.
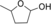
AOX* showed activity towards the shortest tested diol (1,3-propanediol), but a mixture of products was obtained. The enzyme is likely to convert this substrate into a very active aldehyde and/or dialdehyde species. The substrate 1 (1,4-butanediol) was oxidized by AOX* into a mixture of two products: the corresponding γ-butyrolactol and γ-butyrolactone (Table 1). The lactol product intermediate was the main component and existed in the buffer environment as mixture of hemiacetal and hydroxylaldehyde (ratio 10:1). The unanticipated finding that AOX* converted 1 into the lactone (by a double oxidation) motivated us to perform product analysis of other aliphatic diols. The substrate 2 was crucial to understanding the reaction mechanism. For 2, complete conversion was observed, resulting in the doubly oxidized product. This result shows that AOX* performs two consecutive oxidations of the primary alcohol group, even in the presence of a secondary alcohol moiety. The product of the double oxidation was obtained as mixture of the lactone form (γ-valerolactone) and the hydrolyzed form (4-hydroxypentanoic acid) in a ratio of 4:1. Analogous to the conversion of 1, the double oxidation of 2 presumably proceeds by formation of the corresponding lactol (5-methyltetrahydrofuran-2-ol). To verify the ability of AOX* to act on both compounds, the steady-state kinetic parameters for these and other alcohols were determined (see Table 2). This data indeed confirmed that AOX* can act on aliphatic diols and the lactol (5-methyltetrahydrofuran-2-ol), demonstrating that the catalytic route proceeds through initial lactol formation and subsequent additional oxidation to the lactone.
Compound number |
Substrate |
Intermediate |
Product |
Product [%] |
|---|---|---|---|---|
1 |
|
|
|
11 (63 intermediate) |
2 |
|
|
|
>99[b] |
3 |
|
|
|
>99 |
4 |
|
|
|
>99 |
5 |
|
|
|
>99 |
6 |
|
|
|
64[c] (36 aldol) |
7 |
|
|
|
>99[c] |
8 |
|
|
|
>99[d] |
9 |
|
|
|
>99 |
10 |
|
– |
|
>99 |
11 |
|
– |
|
21 |
12 |
|
– |
|
15 |
13 |
|
– |
– |
0 |






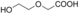
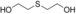

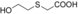


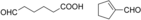




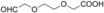






- [a] Reaction conditions: substrate (20 mm), enzyme (40 μm), and 100 mm potassium phosphate buffer pH 7.5 for 48 h at 35 °C. Yields were determined based on 1H NMR spectroscopy. 1H NMR spectroscopy was performed after the addition of D2O (15 % v/v). [b] Ratio cyclic to open product 4:1. [c] Ratio of gem-diol and aldehyde form of carboxylic acid (ratio ≈1:1). [d] Ratio of gem-diol and aldehyde form of carboxylic acid (ratio ≈10:1).
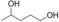
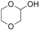
The substrates 3, 4, and 5 were selected as examples for 1,5-diols (Table 1). For all three substrates, complete conversion was obtained and the corresponding products of the double oxidation for these diols were obtained in the form of 5-hydroxycarboxylic acids as single products. No other products (lactone, diacid, or aldehyde acid) were observed by 1H NMR spectroscopy. These results are in contrast to the products formed with 1 and 2, which are obtained in the lactone form. Additional experiments were performed to explain this apparent difference. We investigated the stability of the commercially available 1,4-dioxan-2-one (the lactone form of the product of 4). This compound was found to swiftly hydrolyze in buffer (Scheme 1). In buffer, in less than half hour it completely hydrolyzed to the hydroxy acid (reaction conditions: 100 mm KPi buffer pH 7.5; see the Supporting Information), indicating that as soon as the lactone is formed upon oxidation of either 3, 4, or 5, the formed lactone rapidly hydrolyzes into the stable hydroxy acid. We also monitored the conversion of 4, thus revealing that the first detectable intermediate exists in its hemiacetal form. According to these data, we conclude that the AOX*-catalyzed double oxidation of aliphatic diols proceeds by formation of the corresponding hemiacetal with subsequent oxidation into the lactone, which is prone to hydrolysis into the corresponding 5-hydroxy acids (Scheme 1).

Catalytic route for oxidation of the 1,5-diols 3, 4 and 5 by AOX*. X=C, S, or O.
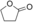
The substrate 6 (Table 1) was also tested because the corresponding product lactone (ϵ-caprolactone) is of value as a polymer building block.20 Somewhat unforeseen, AOX* was found to oxidize both hydroxy groups into aldehyde groups, resulting in the production of adipaldehyde. In the employed buffer, adipaldehyde spontaneously reacted by aldol condensation (non-enzymatic reaction; see Supporting Information), while further oxidation into 6-oxohexanoic acid was observed. Similarly to substrate 6, 1,8-octanediol (7) and triethylene glycol (8) underwent oxidation at both hydroxy groups, yielding the corresponding dialdehydes, which were then further oxidized into oxocarboxylic acids. For these two substrates the aldol condensation product was not obtained, probably because of the unfavorable formation of a seven-membered-ring product. For the latter substrates (6–8), AOX* performed a triple oxidation by oxidizing the gem-diol form of the formed dialdehydes. NMR analysis also confirmed that these aliphatic aldehydes are significantly hydrated (10–50 %) in the employed buffer. For the selective oxidation of only one hydroxy group, as observed for 3–5, formation of a very stable hemiacetal intermediate seems to be crucial. If the hemiacetal is not formed upon the first oxidation, the enzyme will oxidize the other hydroxy group, resulting in the dialdehyde. Subsequently, one aldehyde group is oxidized to the carboxylic acid via the gem-diol. Once that the carboxylic acid is obtained, the enzyme does not accept this compound for further oxidation towards a diacid. Along these lines, the substrate 9 was found to yield hexanoic acid, supporting our hypothesis that AOX* can further oxidize the initially formed aldehyde via its gem-diol. Different from the other substrates, 9 and the intermediate product formed from 9 (Int-9) exhibited substrate inhibition (see Table 2; see the Supporting Information), which may be due to alternative binding pockets for these relatively apolar substrates when compared with the tested diols.
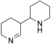
The aminoalcohol 10 was used to explore the substrate promiscuity of AOX* (Table 1). Interestingly, complete conversion was observed for 10. From the 1H NMR spectra it can be concluded that the reaction occurred as selective single oxidation on the hydroxy group. A singlet at δ=7.88 ppm clearly indicated that the product exists as an imine (in situ formed spontaneously from aminoaldehyde). The 1H NMR spectra appeared complex and the product could not be extracted with ethyl acetate to confirm the product by GC-MS analysis. Nevertheless Braekman and co-worker describe that Δ-piperideine, under reaction conditions (pH 7.5) analogous to ours, preferably dimerizes to form tetrahydroanabasine, which correlates with our 1H NMR spectra.21
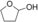
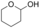
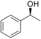
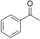
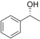
It is known that alcohol oxidases can oxidize secondary alcohols, though with poor efficiency. We selected just a few to test AOX* (substrates 11, 12, and 13; Table 1). Cyclohexanol (11) was oxidized to cyclohexanone with a low yield. Initially, we tested racemic 1-phenylethanol and its conversion was rather low, similar to cyclohexanol. We then tested separately each enantiomer of this alcohol (substrates 12 and 13). Remarkably, AOX* was highly selective towards the S enantiomer of 1-phenylethanol (no conversion for the R enantiomer).
The steady-state kinetic parameters for a selection of the discovered AOX* substrates were determined (Table 2). The observed values for 2 and its corresponding lactol (Int-2) support the proposed catalytic mechanism of the double oxidation going through the lactol intermediate. The rate-limiting step (lower kcat/KM) seems to be the second oxidation step, the oxidation of the lactol. This step was confirmed for 1 and also by shorter conversion experiments for 2–5. Shorter incubations revealed the accumulation of the respective lactols.
Compound number |
Substrate |
KM[a] |
KI[b] |
kcat[a] |
kcat/KM |
|---|---|---|---|---|---|
1 |
|
198 |
n.d. |
6.74 |
34.1 |
2 |
|
52.6 |
n.d. |
1.28 |
24.3 |
Int-2 |
|
95.5 |
n.d. |
1.13 |
11.8 |
3 |
|
69.6 |
n.d. |
3.48 |
50.0 |
4 |
|
12.3 |
n.d. |
0.73 |
59.3 |
5 |
|
119 |
n.d. |
3.56 |
29.9 |
6 |
|
12.9 |
n.d. |
4.07 |
316 |
9 |
|
3.0 |
56 |
0.56 |
187 |
Int-9 |
|
0.20 |
5.5 |
0.15 |
750 |



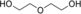
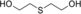



- [a] Values obtained using the HRP-coupled assay in 50 mm potassium phosphate, pH 7.5. KM values are presented in units of mm, kcat values are presented in units of s−1, and kcat/KM values are presented in units of m−1 s−1. [b] The notation n.d. (not detected) used to indicate that no substrate inhibition was observed.
The substrates 4 and 5 were also tested as substrates with several other flavoprotein alcohol oxidases: alditol oxidase (HotAldO) from Acidothermus cellulolyticus 11B, chitooligosaccharide oxidase (ChitO) from Fusarium graminearum, 5-hydroxymethylfurfural oxidase (HMFO) wild-type and variant 8BxHMFO from Methylovorus sp. strain MP688, methanol oxidase from Hansenula sp. (EC 1.1.3.13), glucose oxidase form Aspergillus niger (EC 1.1.3.4), and choline oxidase wild-type and an engineered choline oxidase variant (AcCO6) from Arthrobacter chlorophenolicus.16, 17, 22-24 Except for (AcCO6), all these oxidases did not convert 4 and 5. The sixfold mutant of choline oxidase, AcCO6, proved to facilitate the double oxidation of 4 and 5, albeit with low yields: 5 % product (38 % lactol intermediate) with 4, and 28 % of product (68 % lactol intermediate) with 5, using the same reaction conditions as with AOX*. Furthermore, AcCO6 was found to suffer from severe substrate inhibition.
In conclusion, this work demonstrates the potential of AOX* as a biocatalyst to produce, in one pot, hydroxy acids from 1,5-diols through a selective double oxidation. Diethylene glycol and thiodiglycol can be converted into the corresponding hydroxy acids, which represent interesting building blocks for biodegradable polymers. Similarly, 1,4-diols are converted into the respective lactones. The final oxidation products obtained, γ-butyrolactone and γ-valerolactone, are used in the polymer industry.4, 25 The catalytic mechanism of AOX* with these diols involves the in situ formation of stable hemiacetals. Longer diols can also be oxidized by AOX*, resulting in the corresponding oxocarboxylic acids through a triple oxidation. For all these AOX*-catalyzed oxidation cascade reactions, no external cofactor is required. These results suggest that AOX* holds a great promise as a biocatalyst for selective oxidations.
Experimental Section
AOX* was expressed in E. coli NEB 10β as His-tag-SUMO phusion using a pBAD expression vector and purified using affinity chromatography as described before.19 Kinetic parameters were determined using a HRP-coupled assay as described before.19 1H NMR spectra were recorded on an Agilent 400-MR spectrometer (1H and 13C resonances at 400 MHz and 100 MHz, respectively). Chemical shifts are reported in parts per million (ppm) and coupling constants (J) are reported in hertz (Hz).
Acknowledgements
M.W.F. and C.M. received funding from the Dutch research council NWO (VICI grant).
Conflict of interest
The authors declare no conflict of interest.

