

Elektrochemische Homo- und Kreuzcyclisierung von Alkinen und Nitrilen für die regio- und chemoselektive Synthese von 3,6-Diarylpyridinen
Abstract
Wir berichten eine elektrochemische [2+2+2]-Cyclisierung von Alkinen mit Nitrilen, bei der in einem einzigen Schritt aus kostengünstigen, leicht verfügbaren Ausgangsmaterialien substituierte Pyridine gebildet werden. Durch die Kombination von Elektrochemie und einem Triarylamin-Redoxvermittler kann auf Übergangsmetalle und zusätzliche Oxidationsmittel verzichtet werden. Neben der Bildung von Diarylpyridinen über die Homokupplung zweier identischer Alkine ist auch die Heterokupplung zweier unterschiedlicher Alkine in Abhängigkeit von ihren elektronischen Eigenschaften möglich, was die beispiellose Kontrolle der Chemoselektivität in diesem katalytischen [2+2+2]-Prozess unterstreicht. Mechanistische Untersuchungen wie Cyclovoltammetrie und Crossover-Experimente in Verbindung mit DFT-Berechnungen deuten auf die einleitende Oxidation eines Alkins als Schlüsselschritt hin, der zur Bildung eines Vinylradikalkation führt. Der Einsatz der Durchflusstechnik erwies sich als entscheidend für ein effizientes Scale-up des Prozesses. Die Nützlichkeit der Produkte wird durch die Synthese von Molekülen mit ausgedehnten π-Systemen veranschaulicht, die für die Material- oder Arzneimittelsynthese relevant sind.
Einleitung
Die Allgegenwart der Pyridineinheit in Naturstoffen, Arzneimitteln, organischen Materialien und Katalysatoren unterstreicht ihren privilegierten Charakter; Beispiele, die für diese Arbeit relevant sind, sind in Abbildung 1A dargestellt.1, 2, 3, 4, 5, 6 Angesichts dieser Bedeutung wurden viele metallkatalysierte Wege7 wie C−H-Aktivierung,8 Umlagerung,9 Ringerweiterung,10 Mehrkomponentenreaktionen11 und molekulare Editierverfahren12 für ihre Synthese entwickelt. Übergangsmetall-katalysierte intramolekulare [2+2+2]-Cyclisierungen13 durch Kopplung von zwei Alkinen mit Nitrilen, die den Vorteil der Verwendung von verbundenen Diinen (Abbildung 1B) zur Verringerung des entropischen Nachteils nutzen, haben sich als effektiver Weg zu Pyridinen erwiesen.14 Seit der Pionierarbeit von Wakatsuki und Yamazaki über CpCo(I)-katalysierte Reaktionen15 wurde diese Cycloaddition mit Hilfe verschiedener Übergangsmetallkatalysatoren, darunter Kobalt,16 Nickel,17 Eisen,18 Ruthenium,19 Rhodium,20 und Iridium,21 erforscht. Die intramolekulare Cyclisierung von zwei verschiedenen Alkinen kann ebenfalls erreicht werden, indem entweder die beiden Alkine oder ein Alkin mit dem Nitrilrest verbunden werden17b, 18, 20, 21a, 22 (Abbildung 1B), wodurch die Chemoselektivität kontrolliert werden kann.

Synthese von polysubstituierten Pyridinen: Literaturübersicht und aktuelle Arbeit.
Intermolekulare Übergangsmetall-katalysierte Cycloadditionen zwischen Nitrilen und Alkinen stellen ebenfalls einen einfachen, aber kaum erforschten atomökonomischen Ansatz zur Synthese multisubstituierter Pyridine dar (Abbildung 1B)16c, 23. Allerdings war bei dieser Strategie nur die Kopplung zweier identischer und symmetrischer Alkine möglich, während unsymmetrische, z. B. endständige Alkine in der Regel zu Regioisomeren führen. Nach unserem Kenntnisstand wurde die intermolekulare Kopplung zwischen zwei verschiedenen Alkinen und einem Nitril nur schrittweise unter Verwendung stöchiometrischer Mengen von Cp2ZrEt2 und NiCl2(PPh3)2 von Takahashi und Mitarbeitern24a oder unter Verwendung von Titanalkoxid von Sato und Mitarbeitern24b durchgeführt, alternativ wurden Vinyliodoniumsalze als ein Kopplungspartner eingesetzt.24c
Eine katalytische Strategie zur Realisierung einer Dreikomponenten-Kreuzcyclisierung von zwei verschiedenen Alkinen mit Nitrilen würde daher eine wertvolle Erweiterung des derzeitigen Stands der Technik darstellen und die Synthese verschiedener Pyridingerüste ermöglichen.
Jüngste Entdeckungen und ein Aufschwung der Forschung im Bereich der Photo- und Elektrochemie bieten eine Alternative zu den klassischen metallvermittelten Reaktionsbedingungen für den Aufbau von Pyridinringen. Wang, Meng und Mitarbeiter berichteten über eine photokatalytische [2+2+2]-Cyclisierung aromatischer Alkine mit Nitrilen zur Synthese von 2,3,6-trisubstituierten Pyridinen unter Verwendung eines Pyryliumsalzes als Photoredox-Katalysator (Abbildung 1C),25 mit dem die hohen Oxidationspotenziale von Alkinen, die die größte Herausforderung bei dieser Umwandlung darstellen, überwunden werden konnten. Die Elektrochemie hat den Vorteil, dass Elektronen als spurlose Redoxreagenzien zur Aktivierung gewöhnlicher und inerter organischer Moleküle, insbesondere Alkine,26 eingesetzt werden können, um hochreaktive Zwischenprodukte zu erzeugen. In jüngster Zeit haben elektrochemische Umwandlungen eine Renaissance erlebt,27 und im Rahmen dieser Studie wurden kürzlich elektrochemische [2+2+2]-Homocyclotrimerisierungen von Alkinen zu Arenen untersucht (Abbildung 1D).28
Wir demonstrieren hier eine intermolekulare Cyclisierungsstrategie von Alkinen und Nitrilen, bei der 2,3,6-trisubstituierte Pyridine durch Elektrolyse (Konstantstrom) unter Verwendung einer Kombination aus einer katalytischen Menge Triphenylamin (TPA) und Benzoesäure als Redox-Shuttle entstehen, die den Einsatz von Metallen und anderen externen Additiven überflüssig macht (Abbildung 1E). Die Kreuzcyclisierung von zwei verschiedenen Alkinen ist insbesondere möglich und nach unserem Kenntnisstand bisher einmalig. Außerdem wird keine konkurrierende Alkin-[2+2+2]-Cyclotrimerisierung beobachtet.
Ergebnisse und Diskussion
Wir nahmen an, dass der Schlüssel zum Erfolg dieser Methode in der Bildung eines elektrophilen Radikalkations durch die Ein-Elektronen-Oxidation von Alkinen liegt (Schema 1). Während die direkte anodische Oxidation von Phenylacetylen (1 a, Eoxp/2=+2.35 V) in Acetonitril (2 a) tatsächlich das gewünschte Cycloaddukt 3 a ergab, war die Ausbeute nur mäßig (Tabelle 1, Eintrag 2). In Anlehnung an den Erfolg von Triarylaminen als Redox-Vermittler in elektrochemischen Reaktionen29 setzten wir katalytisch TPA (8 mol %) ein, um den Prozess zu erleichtern (siehe mechanistische Diskussion, vide infra), wodurch sich die Ausbeute von 3 a verdoppelte (Tabelle 1, Eintrag 1). TPA zusammen mit Tetrabutylammoniumhexafluorophosphat (nBu4NPF6, 0.15 M) als Elektrolyt war optimal, im Gegensatz zu anderen getesteten Kombinationen (Tabelle 1, Einträge 3–5). Ebenso wichtig war der Einsatz von Benzoesäure (5 equiv) als Opferreduktionsmittel, das Wasserstoff an der Kathode erzeugte, während sich andere Protonenquellen als weniger wirksam erwiesen (Tabelle 1, Einträge 6–8). Es sei darauf hingewiesen, dass Benzoesäure kostengünstig ist (etwa 10 % des Preises der als überstöchiometrischer Zusatzstoff in der Elektrochemie weit verbreiteten Trifluoressigsäure)30 und eine unbedenkliche Wahl darstellt, die häufig als Lebensmittelzusatzstoff verwendet wird. Darüber hinaus war die Wahl des Elektrodenmaterials entscheidend: Die besten Ergebnisse wurden mit einer Anode aus netzförmigem Kohlenstoff (RVC) - einem mikroporösen Elektrodenmaterial aus Glaskohlenstoff - und einer Platinkathode erzielt. Eine Graphitplatte war als Anode akzeptabel, wenn auch mit einer etwas geringeren Ausbeute, während die Verwendung von Graphitplatten als beide Elektroden keine Reaktion ergab (Tabelle 1, Einträge 9–10). Eine Verkürzung der Reaktionszeit oder eine Erhöhung des Stroms verringerte die Produktausbeute (Tabelle 1, Eintrag 11–12). Experimente mit geteilten Zellen unterstrichen die Vorteile einer ungeteilten Zelle und deren einfache Handhabung (Tabelle 1, Eintrag 13). Bei der Elektrolyse mit konstantem Potenzial, bei der das anodische Potenzial auf +1.3 V eingestellt wurde, wurde kein Produkt erhalten, während 3 a bei einem konstanten Potenzial von +2.5 V in 23 % Ausbeute gewonnen wurde (Tabelle 1, Eintrag 14). Daraus schlossen wir, dass die anodische Oxidation des Alkins für die Einleitung der Cycloaddition unerlässlich ist. Eine inerte Atmosphäre war wichtig (Tabelle 1, Eintrag 15), und außerdem schloss die fehlende Produktbildung ohne Anlegung von Strom die Möglichkeit einer Hintergrundreaktion aus (Eintrag 16).

TPA-PhCOOH-unterstützte elektrosynthetische [2+2+2]-Cyclisierung von Alkinen mit Nitrilen: Arbeitshypothese.
|
||
Eintrag |
Abweichung von den Standardbedingungen |
Ausbeute 3a (%)[b] |
|---|---|---|
1 |
Keine |
76 (74)[c] |
2 |
ohne Triphenylamin (TPA) |
38 |
3 |
Tris(4-bromphenyl)amin anstelle von Triphenylamin |
17 |
4 |
nBu4NBF4 anstelle von nBu4N ⋅ PF6 |
42 |
5 |
nBu4NPF6 (0.2 M) |
56 |
6 |
ohne PhCOOH |
0 |
7 |
CF3COOH anstelle von PhCOOH |
<5 |
8 |
PivOH anstelle von PhCOOH |
48 |
9 |
Gf(+)/Pt(−) anstelle von RVC(+)/Pt(−) |
66 |
10 |
Gf(+)/Gf(−) anstelle von RVC(+)/Pt(−) |
0 |
11 |
5 mA, 6 h anstelle von 5 mA, 12 h |
47 |
12 |
10 mA, 6 h anstelle von 5 mA, 12 h |
33 |
13 |
Geteilte Zelle (Anode: 1 a, TPA, 0.15 M nBu4NPF6, 4 mL MeCN; Kathode: PhCOOH, 0.15 M nBu4N ⋅ PF6, 4 mL MeCN) |
47 |
14 |
Konstantes Potenzial: @1.3 V/@2.5 V |
0/23 |
15 |
unter Luft |
49 |
16 |
ohne Strom |
0 |
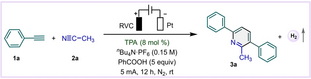
- [a] Reaktionsbedingungen: 1 a (0.5 mmol, 1 equiv), 2 a (4 mL), TPA (8 mol %), nBu4N ⋅ PF6 (0.15 M in 4 mL 2 a), PhCOOH (5 equiv), RVC Anode (1.5 cm×0.8 cm×0.5 cm), Pt-Draht Kathode, Konstanter Strom 5 mA, 12 h, N2, Raumtemperatur, ungeteilte Zelle. [b] Ausbeuten wurden mittles 1H NMR-Spektroskopie bestimmt. [c] Ausbeute des isolierten Produkts.
Mit den optimierten Bedingungen untersuchten wir die Anwendungsbreite der Reaktion (Schema 2). Phenylacetylene mit mäßig elektronenschiebenden (Alkyl) und elektronenziehenden (ortho-F, para-Cl, Br) Gruppen ermöglichten die Kopplung mit 2 a, wodurch 3 b–i in guten bis hohen (51–78 %, Schema 2a) Ausbeuten entstand. Die Struktur von Pyridin 3 i, das sich für weitere Transformationen als wertvoll erwies (vide infra, Schema 4), wurde durch Röntgenkristallanalyse bestätigt.31 Ein Phenylacetylen mit einer stark elektronenabgebenden Gruppe an der para-Position (p-OMe) wurde ebenfalls erfolgreich gekuppelt, allerdings konnte in diesem Fall Pyridin 3 j ohne den TPA-Zusatz erhalten werden. Alkine mit stärkeren elektronenziehenden Substituenten (4-Carbonsäureester, 4-Trifluormethyl, 3,5-disubstituierte Halogenide (F, Cl)) erwiesen sich als nicht reaktiv (3 k–3 n), was darauf hindeutet, dass ihre anfängliche elektrochemische Oxidation zu einem Radikalkation unter diesen Bedingungen nicht möglich ist (vgl. mechanistische Diskussion, vide infra).

Substratspektrum der elektrochemischen [2+2+2]-Cyclisierung zwischen EoAs und Nitrilen. Reaktionsbedingungen: [a] 1 (0.5 mmol, 1 equiv), 2 (4 mL), TPA (8 mol %), nBu4NPF6 (0.15 M in 4 mL 2 a), PhCOOH (5 equiv), RVC Anode (1.5 cm×0.8 cm×0.5 cm), Pt-Draht Kathode (0.06 cm), konstanter Strom=5 mA, 12 h, unter N2, Raumtemperatur, ungeteilte Zelle. [b] ohne TPA, [c] 1 (0.5 mmol, 1 equiv), 2 (10 mmol, 20 equiv), TPA (8 mol %), CH3NO2 (4 mL), nBu4NPF6 (0.15 M in 4 mL CH3NO2), PhCOOH (5 equiv), RVC Anode (1.5 cm×0.8 cm×0.5 cm), Pt-Draht Kathode (0.06 cm), konstanter Strom=5 mA, 12 h, unter N2, Raumtemperatur, ungeteilte Zelle; Isolierte Ausbeuten.
Um die Anwendungsbreite auf der Seite der Nitrile (Schema 2b) auszuloten, musste ein anderes Lösungsmittel als Acetonitril gefunden werden. Nach einem umfangreichen Screening (Details siehe S.I.; Tabellen S10 und S11) erwies sich Nitromethan für die Kopplung von aliphatischen Nitrilen mit unterschiedlichen Kohlenstoffketten mit Phenylacetylenen als geeignet, um die entsprechenden 2,3,6-trisubstituierten Pyridine (3 o–3 t, Schema 2b) zu erhalten. Nitrile mit einem Halogenatom an der Endposition waren ebenfalls erfolgreiche Kupplungspartner und lieferten die Pyridinderivate 3 u, 3 v (bestätigt durch Röntgenstrukturanalyse)31 und 3 w; bei letzterem wurde jedoch eine kompetitive Reduktion zu 3 a beobachtet. Nitrile mit einer vinylischen und allylischen Doppelbindung wurden in die Pyridinderivate 3 x und 3 y umgewandelt. Die geringeren Ausbeuten könnten durch eine konkurrierende radikalische Cyclisierung des Aren-Zwischenprodukts an der Alken-Doppelbindung erklärt werden, und tatsächlich wurde ein solcher Prozess durch die Isolierung von 3 x′, das zusammen mit 3 x gebildet wurde, bestätigt. Deuteriertes Acetonitril (CD3CN) lieferte d3-3 a in guter Ausbeute, ohne dass eine Erosion des eingebauten Deuteriums zu beobachten war. Bei der Verwendung von aromatischen Nitrilen wurde jedoch keine Produktbildung beobachtet.
Um die Tatsache zu einen Vorteil umzumünzen, dass elektronenarme Alkine (EnoA) nicht in der Titelreaktion reagieren, untersuchten wir, ob die Kopplung von Nitrilen mit zwei elektronisch unterschiedlichen Alkinen (EoA und EnoA) möglich ist. In der Tat wurde durch die Kombination von 1 a (1 equiv), das bei der Homo-Cyclisierung erfolgreich war, mit dem erfolglosen Alkin 1 k (1.3 equiv) das kreuzcyclisierte Produkt 4 a in 64 % Ausbeute (Schema 3A) zusammen mit 8 % des homocyclisierten Produkts 3 a erhalten, jedoch wurde kein homo-gekoppeltes Produkt 3 k beobachtet. Folglich kombinierten wir erfolgreich 1 a mit den Alkinen 1 l–o zu den kreuzcyclisierten Produkte 4 b–4 e (Schema 3B), was durch eine Röntgenstrukturanalyse von 4 b und 4 j eindeutig nachgewiesen werden konnte.32 Das aus 4-Bromphenylacetylen 1 i (EoA) und 3,5-Dibromphenylacetylen 1 p (EnoA) synthetisierte Pyridinderivat 4 g (53 % Ausbeute) zeigt, dass subtile Unterschiede in der elektronischen Natur der Alkine ausreichen, um Heterokupplungen zu erreichen.

Elektrochemische Dreikomponenten-[2+2+2]-Cyclisierung von EoAs, EnoAs und Nitrilen. Reaktionsbedingungen: [a]1 (0.25 mmol, 1 equiv), 2 a (4 mL), 1 k–t (0.325 mmol, 1.3 equiv), TPA (8 mol %), nBu4NPF6 (0.15 M in 4 mL 2 a), PhCOOH (5 equiv), RVC Anode (1.5 cm×0.8 cm×0.5 cm), Pt-Draht Kathode (0.06 cm), konstanter Strom=5 mA, 12 h, unter N2, Raumtemperatur, ungeteilte Zelle. [b] 2 b (10 mmol, 20 equiv), CH3NO2 (4 mL); Isolierte Ausbeuten.
Darüber hinaus lieferte die Funktionalisierung bioaktiver Moleküle und Agrochemikalien unter Verwendung von 4-Ester-substituierten Phenylacetylenen, die von (−)-Menthol, (±)-Isoborneol und Methylparaben abgeleitet sind, die gewünschten Pyridinderivate 4 h–j in mäßigen bis guten Ausbeuten (Schema 3B).
Ein Scale-up im Batch-Verfahren wurde für die Kopplung von 1 i (6 mmol) und 2 a (Schema 4A) demonstriert, wobei eine Erhöhung des Stroms auf 12 mA und eine Verlängerung der Reaktionszeit auf 72 h erforderlich waren, um 3 i in 66 % Ausbeute zu erhalten. Falls gewünscht, kann ein Teil der Benzoesäure (40 %) während der Aufarbeitung zurückgewonnen werden (siehe S.I. für Einzelheiten).

Synthese im Gramm-Maßstab im Batch- und Durchflussverfahren und synthetische Anwendungen.
Die Umwandlung konnte auch in einem kontinuierlichen Durchflussreaktor durchgeführt werden: Im Vergleich zum Batch-Setup war die Reaktionszeit bei ähnlicher Effizienz deutlich kürzer (1 mmol Maßstab, 16 h, 69 % Ausbeute; tR=20 min pro Durchgang vs. 72 h, 66 % Ausbeute; Schema 4B, siehe S.I. für Details). Eine Ansatzvergrößerung auf 8 mmol erhöhte die Reaktionszeit auf 24 h (56 % Ausbeute von 3 i) und erwies sich als robust und reproduzierbar.
3 i bildete die Grundlage für die Untersuchung von Funktionalisierungen (Schema 4b). Mit dem Ziel, das π-System zu erweitern, führte eine Suzuki–Miyaura-Reaktion zu 5 (82 % Ausbeute), während die Abfolge von Sonogashira-Kupplung, TMS-Schützung und kupferkatalysierter Azid-Alkin-Cycloaddition das Bis-Triazolyl-Derivat 7 ergab (59 % Gesamtausbeute über drei Schritte).
Mechanistischen Untersuchungen begannen mit einer Kontrollreaktion, bei der das unabhängig hergestellte TPA-Radikalkation direkt anstelle von TPA eingesetzt wurde (Abbildung 2A, siehe S.I. für Einzelheiten): Die Kopplung von 1 a mit 2 a ergab das gewünschte Produkt 3 a in ähnlicher Ausbeute (67 %), aber in einer verkürzten Reaktionszeit von 8 statt 12 h. Eine kontinuierliche Produktbildung wurde von Anfang an beobachtet, während unter den Standardreaktionsbedingungen eine anfängliche unproduktive Phase zu beobachten ist, die vermutlich für eine vollständige anodische Oxidation des leichter zu oxidierenden TPA erforderlich ist, bevor das Alkin oxidiert werden kann. Wurden dem Reaktionsgemisch TEMPO oder BHT als Radikalfänger zugesetzt (Abbildung 2B), wurde die Reaktion vollständig gehemmt, was mit einem Radikalmechanismus übereinstimmt. An der Kathode wurde eine Wasserstoffgasentwicklung festgestellt, die qualitativ durch GC analysiert (Abbildung 2C, siehe S.I. für Einzelheiten) oder für die Hydrierung von 1,2-Dihydronapthalin (siehe S.I. für Einzelheiten.) verwendet wurde.

Mechanistische Studien und plausibler Mechanismus.
CV-Messungen (siehe S.I. für Einzelheiten) ergaben, dass das Oxidationspotenzial von TPA +0.99 V gegenüber SCE beträgt, während das von Arylacetylenen über +2.1 V gegenüber SCE liegt. Somit sollte TPA zunächst an der Anode vollständig oxidiert werden, aber aufgrund der unterschiedlichen Potenziale ist es unwahrscheinlich, dass es als Vermittler29, 33 für die Oxidation der EoAs fungiert. Wir schlagen daher vor, dass die EoAs nach der vollständigen Oxidation von TPA eine direkte Einzelelektronenoxidation an der Anode erfahren. Wir haben dann die Rolle von TPA hinterfragt, das zwar für die Reaktivität nicht notwendig, aber für eine verbesserte Ausbeute von Vorteil ist (Tabelle 1, Einträge 1 und 2). CV–Messungen von TPA-PhCOOH-Gemischen (Abbildung 2D) zeigen, dass mit zunehmendem Molverhältnis von Benzoesäure die Oxidationsstromstärke von TPA stetig abnimmt, während die Reduktionsstromstärke von PhCOOH allmählich zunimmt. Dies bedeutet, dass die für die Oxidation verfügbare „frische“ TPA-Konzentration an der Elektrode mit zunehmender PhCOOH-Beladung abnimmt. EPR-Messungen zeigen das vollständige Verschwinden des starken Signals des TPA-Radikalkations34 nach dem Mischen mit Kaliumbenzoat (Abbildung 2E, siehe S.I. für Einzelheiten), was auf die Ein-Elektronen-Reduktion des TPA-Radikalkations durch das Benzoat-Anion schließen lässt.
Ausgehend von den obigen Ausführungen beginnt eine plausible mechanistische Erklärung (Abbildung 2F) mit der anfänglichen elektrochemischen Oxidation von TPA, um das TPA-Radikalkation bei einem relativ milden Potenzial (1.0 V gegen SCE) zu bilden. Die Reduktion der Benzoesäure wiederum führt zur Wasserstoffgasentwicklung und zur Bildung eines Benzoatanions (BA−), das mit dem TPA-Radikalkation einen Ladungstransferkomplex A bildet. In der produktiven Phase der Reaktion wird aus EoA 1 an der Anode durch eine Einelektronenoxidation ein Vinylradikalkation I erzeugt. Durch nucleophilen Abfang von I durch Nitril 2 entsteht das Zwischenprodukt II, das vorzugsweise mit EnoA eine Cycloaddition eingeht und das Vinylradikal-Zwischenprodukt III/IV erzeugt. Die Übertragung eines Elektrons aus dem Ladungstransferkomplex A mit gleichzeitiger Protonierung35 liefert das protonierte, quervernetzte Produkt 4-H+ mit gleichzeitiger Bildung eines Phenylcarbonsäure-Radikals (BA⋅). Bei der Reduktion des letzteren an der Kathode entsteht ein Phenylcarboxylat-Anion (BA−), das mit dem TPA-Radikalkation zu A rekombiniert und für den nächsten Zyklus bereit steht. Das Zwischenprodukt IV kann auch direkt an der Kathode reduziert werden, wobei wir davon ausgehen, dass dies weniger effektiv ist als SET durch Komplex A.
Um die kontraintuitive Bevorzugung zweier elektronenarmer Komponenten (II und EnOA), die selektiv miteinander wechselwirken und so die Kreuzkupplung ermöglichen, zu verstehen, wurden die Grenzmolekülorbitale der reagierenden Partner II-1 a–2 a und 1 a, 1 k und 1 p analysiert (Abbildung 3).

FMO-Analyse zur Erklärung der Notwendigkeit elektronenarmer Alkine und der beobachteten Chemoselektivität, d. h. der Bevorzugung von 1 p gegenüber 1 a bei der Reaktion mit II-1 a–2 a.
Aufgrund der Orbitalenergien (ϵ) ist nur eine Wechselwirkung zwischen dem LUMO des Radikalkations II-1 a–2 a und dem HOMO des Alkins 1 möglich. Daher lässt sich die Kopplung zwischen II und 1 am besten als formale [4+2]-Cycloaddition36 der C−N- und C−C-π-Bindungen ohne Beteiligung des Radikalzentrums beschreiben. Um energetisch mit dem niedrig liegenden LUMO des Radikalkations II zusammenzupassen, muss das HOMO des Alkins niedrig genug sein, was eine Akzeptorsubstitution am Alkin begünstigt. Ein gutes Indiz dafür, dass die mechanistische Hypothese einer formalen [4+2] Cycloaddition eine geeignete Beschreibung ist, zeigt die Beobachtung, dass die Grenzorbtial-Abstände (Δϵ) eine Vorhersagekraft für die Substratselektivität haben. Die Korrelation zwischen den Δϵ-Werten und der experimentell beobachteten Bevorzugung von II-1 a–2 a für elektronenarme Alkine wie 1 k und 1 p gegenüber 1 a lässt sich mit dem geringeren FMO-Abstand erklären, der eine abnehmende Barriere für diesen Elementarschritt impliziert.
Die Vorzeichen der wechselwirkenden Orbitale (Abbildung 3A) deuten darauf hin, dass ein konzertierter Weg möglich ist, der zu einem stark resonanzstabilisierten Radikalkation III führt, das im Vergleich zu den Ausgangsstrukturen energetisch deutlich niedriger liegt (ΔRH=−110.1 kcal/mol für II-1 a–2 a+1 p). Dieser konzertierte Weg erklärt auch die Regioselektivität für das postulierte 1,2,5-substituierte III, welches das ungepaarte Elektron über den Arylring des ehemaligen Alkinreaktanden delokalisieren kann, während das entgegengesetzte Regioisomer diese Möglichkeit nicht hat (siehe S.I. für Einzelheiten).
Schlussfolgerung
Zusammenfassend haben wir eine einfache und skalierbare Methode entwickelt, um aus handelsüblichen Ausgangsstoffen trisubstituierte Pyridine durch elektrochemisch induzierte [2+2+2]-Annulation aromatischer Alkine mit Nitrilen zu bilden. Insbesondere haben wir zwei Klassen von Alkinen (EoAs und EnoAs) identifiziert, die sich in ihrer Fähigkeit unterscheiden, elektrochemisch oxidiert werden zu können. Wir haben sie erfolgreich mit Nitrilen in einer intermolekularen Dreikomponenten-Kreuzcyclisierung eingesetzt, die die erste ihrer Art ist. Darüber hinaus hat die Skalierbarkeit in Batch- und Durchflussverfahren die Effizienz des entwickelten Protokolls bewiesen. Die synthetische Diversifizierung zu π-verlängerten Aromaten unterstreicht den Nutzen dieser Methode. Es wurde ein plausibler Mechanismus vorgeschlagen, der durch eine Reihe von mechanistischen Studien, Experimenten zum Radikalabfang, CV-, EPR-, NMR- und Berechnungen gestützt wird. Die hier entwickelte elektrochemische Strategie ist komplementär zu der gut etablierten und metallvermittelten [2+2+2]-Cycloaddition zwischen Alkinen und Nitrilen, zeichnet sich aber durch ihre Fähigkeit aus, Kreuzcyclisierungen mit zwei unterschiedlichen Alkinen mit hoher Chemo- und Regioselektivität zu ermöglichen.
Danksagung
Diese Arbeit wurde von der Deutschen Forschungsgemeinschaft (DFG) – TRR 325444632635-A2 (Doktorandenstipendium MG), der Europäischen Kommission (Marie Curie Action CuII-VLIH – 101066526; Postdoc-Stipendium TM) und dem Elitenetzwerk des Bayerischen Doktorandenkollegs (IDK Chemische Katalyse mit photonischer oder elektrischer Energiezufuhr, Doktorandenstipendium ML). Wir danken Sabine Stempfhuber und Birgit Hischa für Röntgenstrukturanalysen, Regina Hoheisel für Cyclovoltammetrie Messungen und Dr. Gábor Balázs für EPR-Messungen (alle Universität Regensburg). Open Access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.
Interessenkonflikt
Die Autoren erklären, dass kein Interessenkonflikt besteht.
Open Research
Data Availability Statement
Die Daten, die die Ergebnisse dieser Studie untermauern, sind im Zusatzmaterial zu diesem Artikel zu finden.

