

Copper-Catalyzed Regiodivergent Internal Allylic Alkylations
Abstract
We report a copper-catalyzed, regioselective, and stereospecific alkylation of unbiased internal allylic carbonates with functionalized alkyl and aryl Grignard reagents. The reactions exhibit high stereospecificity and regioselectivity for either SN2 or SN2′ products under two sets of copper-catalyzed conditions, which enables the preparation of a broad range of products with E-alkene selectivity. Density functional theory calculations reveal the origins of the regioselectivity based on the different behaviors of homo- and heterocuprates.
Introduction
Nucleophilic substitutions represent one of the most important classes of reactions in organic chemistry. Catalytic reactions between allylic electrophiles and carbon-based nucleophiles are particularly useful because of the synthetic utility of the alkene containing allylic alkylation products.1, 2, 3, 4 Elegant examples of regiocontrol and stereocontrol have been reported for the alkylation of simple terminal allylic electrophiles (Scheme 1A), including enantiospecific processes governed by stereoelectronic effects and enantioselective reactions controlled by chiral catalysts. Nevertheless, the origin of regioselectivity for SN2 or SN2′ product formation in the nucleophilic displacement of complex internal allylic electrophiles remains unsolved for catalytic allylic alkylation chemistry, which has prevented the discovery of more general reaction manifolds.

Strategies for catalytic regioselective allylic alkylation of internal allylic electrophiles. (A) Regiocontrol in the alkylation of simple terminal allylic electrophiles. (B) Regiocontrol in the alkylation of biased internal allylic electrophiles. (C) Challenge of alkylation of unbiased internal allylic electrophiles. (D) Our method for the regiodivergent and selective alkylation of unbiased internal allylic electrophiles.
Currently, the most effective catalytic methods for regioselective alkylation of complex internal allylic electrophiles rely on the use of specialized nucleophiles, such as electron-deficient arenes, aryl boronic acids, silyl ketene acetals, or alkynes.5-17 We were interested in employing a broad class of carbon-based nucleophiles, such as Grignard reagents, in regioselective allylic alkylations.
Significant advances in regioselectivity have been made in the copper-catalyzed substitution of allylic electrophiles that are sterically or electronically biased (Scheme 1B).18-21 Most allylic alkylations with Grignard reagents of unbiased internal allylic electrophiles result in the formation of constitutional isomers as well as alkene isomers, due to the unbiased allylic system generated after leaving group dissociation (Scheme 1C).22, 23 Herein, we report a general catalyst-controlled allylic alkylation of unbiased allylic carbonates with aryl and aliphatic Grignard reagents in high regioselectivity and E-alkene selectivity (Scheme 1D). We demonstrate the selective formation of either SN2 or SN2′ products under two sets of copper-catalyzed conditions with stereospecific product formation. Furthermore, density functional theory (DFT) calculations provide mechanistic information and account for the origin of catalyst-controlled regioselectivity, which will provide insight for the development of other classes of regioselective transformations.
Results and Discussion
Optimization Study and Reaction Development
Our studies commenced with the copper-catalyzed coupling of allylic substrates derived from dec-6-en-5-ol and ethylmagnesium bromide (Table 1). In the presence of 10 mol % CuBr ⋅ SMe2 with CH2Cl2 as the solvent, (E)-dec-6-en-5-yl acetate (E-1 a) and (E)-dec-6-en-5-yl methyl carbonate (E-1 b) reacted with ethylmagnesium bromide to furnish coupled products in 12 % and 44 % yield, respectively, as a mixture of constitutional and alkene isomers 2, 3, and 5 (entries 1–2). While both substrates favored the formation of E-alkene isomers 2 and 3, no regioselectivity was observed for SN2′ (2) vs. SN2 (3) pathways. Given the superior yield of product formation with (E)-dec-6-en-5-yl methyl carbonate E-1 b (entry 2), the carbonate-derived substrates were selected for further examination. In the presence of 10 mol % of various copper salts and ethylmagnesium bromide, allylic carbonate E-1 b furnished coupled products 2, 3, and 5 in varying yields and product ratios (entries 2–6). Most notably, CuCN catalyzed the formation of coupled products in 90 % yield with high selectivity for the SN2′ (2 and 5), albeit with no E/Z-alkene selectivity (entry 6).
|
|||||
|
1 |
Catalyst |
Solvent |
Yield[b] |
2 : 3 : 4 : 5[c] |
|---|---|---|---|---|---|
1 |
E-1 a |
CuBr ⋅ SMe2 |
CH2Cl2 |
12 % |
41 : 44:0 : 15 |
2 |
E-1 b |
CuBr ⋅ SMe2 |
CH2Cl2 |
44 % |
42 : 47:0 : 11 |
3 |
E-1 b |
Cu(MeCN)PF6 |
CH2Cl2 |
51 % |
43 : 48:0 : 9 |
4 |
E-1 b |
CuTC |
CH2Cl2 |
42 % |
47 : 53 : 0 : 0 |
5 |
E-1 b |
CuI |
CH2Cl2 |
36 % |
45 : 5:0 : 50 |
6 |
E-1 b |
CuCN |
CH2Cl2 |
90 % |
50 : 0 : 0 : 50 |
7 |
E-1 b |
CuCN |
CHCl3 |
81 % |
48 : 8:0 : 44 |
8 |
E-1 b |
CuCN |
THF |
38 % |
42 : 12:0 : 46 |
9 |
E-1 b |
CuCN |
Et2O |
21 % |
45 : 8:0 : 47 |
10 |
E-1 c |
CuCN |
CH2Cl2 |
82 % |
65 : 0 : 0 : 35 |
11 |
Z-1 c |
CuCN |
Et2O |
80 % |
98 : 1:0 : 1 |
12 |
Z-1 c |
CuCN |
CH2Cl2 |
84 % |
98 : 1:0 : 1 |
13[d] |
Z-1 c |
CuCN |
CH2Cl2 |
76 % |
97 : 1:0 : 2 |
14 |
E-1 c |
Cu(OTf)2 |
CH2Cl2 |
64 % |
41 : 59 : 0 : 0 |
15 |
E-1 c |
Cu(OTf)2 |
Et2O |
70 % |
20 : 80 : 0 : 0 |
16 |
E-1 c |
Cu(OAc)2 |
Et2O |
82 % |
36 : 64 : 0 : 0 |
17 |
E-1 c |
Cu(acac)2 |
Et2O |
82 % |
38 : 62:0 : 9 |
18 |
E-1 c |
CuCl2 |
Et2O |
80 % |
33 : 67 : 0 : 0 |
19 |
E-1 c |
CuBr2 |
Et2O |
85 % |
37 : 63 : 0 : 0 |
20[e] |
E-1 c |
Cu(OTf)2 |
Et2O |
80 % |
15 : 85 : 0 : 0 |
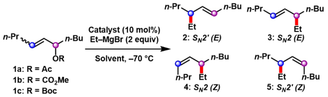
- [a] Reaction conditions: allylic substrate 1 (0.20 mmol), copper catalyst (10 mol %), ethylmagnesium bromide (2 equiv), solvent (0.1 M), −70 °C, 24 h. [b] Isolated yield of allylic alkylation product. [c] Product ratios determined by 13C NMR analysis. [d] CuCN (5 mol %). [e] Terpyridine (12 mol %).
While other reaction solvents did not improve the selectivity for product formation (entries 7–9), a slight increase in selectivity was observed for the E-isomer of the SN2′ pathway (2 : 5=65 : 35) with bulkier (E)-dec-6-en-5-yl t-butyl carbonate (E-1 c, entry 10). Interestingly, by utilizing the Z-isomer of dec-6-en-5-yl t-butyl carbonate (Z-1 c), we obtained the coupled product in 80 % yield in ethereal solvent with high SN2:SN2′ regioselectivity (1 : 99, entry 11) and 84 % yield in CH2Cl2, with the same regioselectivity (entry 12). Optimized conditions were achieved by lowering the catalyst loading of CuCN to 5 mol % while still maintaining the efficiency and selectivity of the transformation, as the alkylation product was formed in 76 % yield with 1 : 99 SN2 : SN2′ regioselectivity and 97 : 2 E/Z-alkene selectivity (entry 13).
Intrigued by the possibility of redirecting the product selectivity to the SN2 pathway, other copper salts that previously resulted in higher yields of SN2 product 3 were studied (entries 2–4). Copper(II) salts were examined with the E-isomer of dec-6-en-5-yl t-butyl carbonate (E-1 c) to determine if the SN2 pathway could be favored. Whereas Cu(OTf)2 catalyzed the formation of coupled products with no regioselectively for SN2 (3) vs. SN2′ (2) pathways in CH2Cl2 (59 : 41, entry 14), a considerable increase in SN2 product formation was observed with Cu(OTf)2 in ethereal solvent (80 : 20, entry 15). While other copper(II) sources did not improve regioselectivity (entries 16–19), we observed a comparable yield (80 %) and increased SN2-regioselectivity (85 : 15) with the addition of catalytic amounts of terpyridine ligand (entry 20). Several other ligands were examined with no further improvement in the regioselectivity (See Supporting Information for details).
With optimized conditions identified for the selective formation of either E-SN2′ product (entry 13) or E-SN2 product (entry 20), the generality of these regiodivergent processes was explored. The scope of products formed through SN2′-selective pathways are summarized in Table 2. Ethylmagnesium bromide was reacted with a series of unbiased internal Z-allylic carbonates in the presence of CuCN in CH2Cl2 to furnish allylic alkylation products in synthetically useful yields with selectivity for the E-SN2′ product (Table 2A). Notably, the method distinguished between similar substituents on the central allylic system, including n-propyl vs. n-butyl (2 a) and n-propyl vs. c-hexyl (2 b). The selective formation of either constitutional isomer 2 b or 2 c from isomeric starting materials demonstrates the regiospecificity of the reaction. High SN2′ selectivity was observed even with substitution at a neopentyl position (2 d).

- [a] Reaction conditions: allylic substrate 1 (0.25 mmol), CuCN (5 mol %), Grignard reagent (1.2 equiv), CH2Cl2 (0.1 M), −70 °C, 40 h. Isolated yield of allylic alkylation product. Product ratios determined by 13C NMR analysis.
Whereas aryl substituted internal allylic substrates often furnish the alkylation products with alkenes in conjugation with the aromatic ring,24 we observed the selective formation of coupled products with the alkene out of conjugation (2 e–2 i). The transformation was tolerant of aromatic rings functionalized with methyl, methoxy, trifluoromethyl, and fluoride substituents. We also generated SN2′ alkylation products containing an aniline (2 j), silyl ether (2 k), and alkyl chloride (2 l).
The SN2′-selective allylic alkylation of (E)-dec-6-en-5-yl t-butyl carbonate (E-1 c) was also examined with several Grignard reagents (Table 2B). The reaction accommodated primary (2 m–2 n), secondary (2 o), tertiary (2 p), benzylic (2 q), and aryl (2 r–2 t) Grignard reagents, with the desired products formed in synthetically useful yields and >90 % selectivity for the E-SN2′ product.
To demonstrate the generality of the SN2-selective pathway, various internal E-allylic substrates and ethylmagnesium bromide were treated with catalytic Cu(OTf)2 and terpyridine in Et2O (Table 3A). Once more, the method distinguished between similar substituents on the central allylic system, including n-propyl vs. n-butyl (3 a) and n-propyl vs. c-hexyl (3 b). A series of styrenyl products (3 c–3 f) were selectively formed from (E)-1-arylhept-1-en-3-yl carbonates. The reaction also tolerated a substrate with an aniline substituent (3 g). Moreover, when subjected to the SN2-selective conditions, (E)-dec-6-en-5-yl t-butyl carbonate reacted with various primary (3 h–3 j), benzylic (3 k), secondary (3 l), and aryl (3 m–3 n) Grignard reagents to yield the corresponding E-SN2 allylic alkylation products (Table 3B). This reaction was also performed on a preparative 1.0 mmol scale (See Supporting Information).

- [a] Reaction conditions: allylic substrate 1 (0.25 mmol), Terpyridine (12 mol %), ethylmagnesium bromide (1.5 equiv), Et2O (0.1 M), −70 °C, 40 h. Isolated yield of allylic alkylation product. Product ratios determined by 13C NMR analysis.
Mechanistic Studies
A series of control experiments were conducted to further understand the origin of regiodivergent alkylation. In a radical trapping experiment, 1.0 equivalent of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) was employed under the standard reaction conditions (Scheme 2A). Despite the large excess of TEMPO compared to the copper catalyst, the catalytic cycle was not inhibited, only impacting the reaction efficiency with reduced yields of 2 a and 3 a to 46 % and 51 %, respectively. We next examined the stereospecificity in the formation of the SN2′ alkylation product 3 o and SN2 alkylation product 3 a from enantioenriched starting materials under standard reaction conditions. For both processes, we observed greater than 90 % enantiomeric specificity with the inversion of configuration (Scheme 2B).

Mechanistic experiments. (A) Radical trapping experiments. (B) Control experiments for stereospecificity. Reaction conditions: (a) allylic substrate 1 (0.25 mmol), CuCN (5 mol %), ethylmagnesium bromide (1.2 equiv), CH2Cl2 (0.1 M), −70 °C, 40 h. (b) allylic substrate 1 (0.25 mmol), Cu(OTf)2 (10 mol %), Terpyridine (12 mol %), ethylmagnesium bromide (1.5 equiv), Et2O (0.1 M), −70 °C, 40 h. Isolated yield of allylic alkylation product. Product ratios determined by 13C NMR analysis.
As the in situ reduction of Cu(OTf)2 in the presence of Grignard reagents was reported by Nishii and co-workers,25, 26 the role of Grignard reagent stoichiometry was investigated in order to identify the oxidation state of the active catalyst. Allylic carbonate Z-1 c was treated with 1.0 equivalent of copper catalyst and 1.0, 2.0, or 3.0 equivalents of ethylmagnesium bromide, respectively (Scheme 3). With CuCN as the catalyst, the results show that the amount of the Grignard reagent did not seem to correlate with the yield of the SN2′ product. In the case of Cu(OTf)2, with 1.0 and 2.0 equivalents of the Grignard reagent, the reaction did not proceed to an appreciable extent. It is noteworthy that 3.0 equivalents of the Grignard reagent generated the alkylation product in 74 % yield. These results strongly suggest that the SN2-selective reaction proceeds via an anionic Cu(l) species, and not a Cu(ll) or neutral organocopper species.26-28

Investigation of active copper species and effect of equivalents of Grignard reagent. Isolated yield of allylic alkylation product. Product ratios determined by 13C NMR analysis.
The proposed mechanisms for the two regiodivergent alkylation processes are summarized in Scheme 4. Both experimental and computational investigations are consistent with the active catalysts being heterocuprate (Cat1) for the SN2′ process and homocuprate (Cat2) for the SN2 process (See Supporting Information for a more detailed analysis of the catalyst structure).29-33 The possibility of syn-elimination in both catalysts can be excluded due to the steric hindrance between the carbonate and copper catalyst. Thus, we mainly calculated the anti-elimination pathway, which is consistent with the experimental stereochemical data. We propose that the heterocuprate prefers a π-complexation with the alkene substrate (A2), while the more electron-rich homocuprate prefers a nucleophilic attack rather than π-complex formation. For the heterocuprate, oxidative addition from the π-complex generates the copper(III) intermediate A3 followed by reductive elimination to furnish the alkylation product 2 a.30, 33-35 In contrast, the homocuprate forms Lewis acid-base adduct B1, and subsequent nucleophilic attack by the cuprate gives copper(III) intermediate B2. Finally, the reductive C−C coupling delivers the desired product 3 a.

Proposed mechanism for copper-catalyzed regiodivergent allylic alkylations.
To test the feasibility of the proposed mechanism, detailed DFT calculations were performed, and entries 11 and 15 were selected as model systems, which do not possess any additional pincer ligands. Geometry optimizations, vibration, and solvation calculations were conducted using the B3LYP-D3/6-31G**/LANL2DZ level of theory.36, 37-40 Single point energies were re-evaluated using the B3LYP-D3/cc-pVTZ(-f)/LACV3P level of theory with the self-consistent reaction field (ϵ=4.24 for diethyl ether) approach41 employed to account for solvation effects (See Supporting Information for details).
Figure 1 summarizes the computed energy profile starting from Z-1 c to the alkylation product 2 a. The reaction begins with the binding of Cat1 with Z-1 c to give the Lewis acid-base adduct A1, which is in equilibrium with π-complex A2.42 Subsequently, oxidative addition via a T-shaped transition state A2-TS delivers the η3-allyl-copper complex A3.35, 43, 44 We also evaluated the direct nucleophilic attack by the heterocuprate A1′-TS, and found that it is not competitive, as it requires a higher energy barrier than A2-TS. At the 16-electron square planar complex A3, the detached leaving group can bind to the copper center intramolecularly to give the 18-electron copper complex A4 and A4’.21, 45 The reductive elimination traversing A4-TS and A4′-TS affords copper complexes A5 and A5′, which upon product release furnish the γ-substituted product (2 a) and α-substituted product (4 a).

Energy profile for the formation of SN2′ product 2 a from Z-1 c and CuCN via heterocuprate Cat1.
Notably, the transition state A4-TS is located at −21.0 kcal mol−1, while A4′-TS is located at −18.6 kcal mol−1, which accounts for the observed SN2′ selectivity for the more relaxed E-olefin product 2 a over a Z-olefin product 4 a. This difference in kinetics can be explained by the relaxation of allylic strain in the preorganized Z-olefin, which is not present in the case of E-olefin. In the product dissociation step, the active catalyst Cat1 is regenerated with the association of the Grignard reagent.
The reaction pathway that is catalyzed by homocuprate Cat2 was also evaluated (Figure 2). In the case of using a copper(II) source, it is reduced to copper(I) to give the homocuprate under the reaction conditions.29, 45 Similar to heterocuprate Cat1, homocuprate Cat2 can promote either π-complexation or nucleophilic attack with E-1 c. In contrast to the heterocuprate case, the reaction catalyzed by homocuprate Cat2 prefers nucleophilic attack rather than formation of the unstable π-complex D1 (see below). The homocuprate Cat2 initially binds to the substrate E-1 c to form a low-energy adduct B1. Nucleophilic attack occurs by the dz2 orbital to yield σ-complex B2.

Energy profile for the formation of SN2 product 3 a with the E-1 c and Cu(OTf)2 via homocuprate Cat2.
Nucleophilic attack via B1-TS has a barrier of 11.7 kcal mol−1, and the product B2 lies at −0.5 kcal mol−1, while the higher barrier of 12.3 kcal mol−1 is observed for the C1-TS. Our calculations suggest that the nucleophilic attack is most likely the rate- and regioselectivity determining step. In both transition states B1-TS and C1-TS, the cuprate adopts a linear geometry with the dz2 orbital as the HOMO. The cuprate nucleophile bearing anionic character prefers to attack the more electrophilic α-carbon rather than the γ-carbon. The small energy difference of the nucleophilic attack step is enough to explain the experimentally observed selectivity (Table 1, entry 20) following the Curtin-Hammett principle (See Supporting Information for detailed analysis). For the subsequent reductive elimination, the resulting SN2 product 3 a is generated via B2-TS in an exergonic reaction of −60.6 kcal mol−1 with a barrier of only 5.8 kcal mol−1.
After elucidating a rationale for product regioselectivity, we investigated the catalytic behavior that determines different reaction pathways. As the only difference between homocuprate and heterocuprate is the cyano group and alkyl group, we concluded that the regioselectivity is determined by the ligand, not by the oxidation state of the copper precatalyst (See Supporting Information for more detailed analysis).35
The relative free energies of key species for π-complex formation are illustrated in Figure 3A. In the heterocuprate case, π-complex A2 is in equilibrium with A1 with an almost negligible energy difference of 0.5 kcal mol−1, while in the homocuprate case, the formation of π-complex D1 from B1 is energetically uphill by 14.9 kcal mol−1.

Comparison between preference of two catalysts for π-complexation.
To understand the energetic difference between π-complexes A2 and D1, distortion-interaction analysis was conducted as described in Figure 3B.46, 47 The complex was divided into the alkene substrate and catalyst fragments. The interaction energy of D1 is 22.3 kcal mol−1 greater than A2, which is not surprising considering that the electron-donating ethyl group makes the metal center electron-rich, strengthening the back-donation character. The difference in distortion energy is 35.3 kcal mol−1 which overrides the difference in interaction energy. Compared to the 6.3 kcal mol−1 difference of substrate distortion, the distortion energies of the catalyst part show a significant difference with the energy of 29.0 kcal mol−1. Therefore, we can conclude that the preference for the π-complexation is determined by the stability of the bent structure of the catalyst.
The distortion-interaction analysis quantitatively shows that the heterocuprate prefers π-complexation as it can easily adopt a bent structure. Thus, it is important to understand why the heterocuprate prefers bending compared to the homocuprate. In this context, the correlation between structural transformation and frontier orbitals of both homo- and heterocuprate was investigated with a model system.48, 49 Figure 4 represents the molecular orbitals of homo- and heterocuprates at both linear and bent geometries. In the linear homocuprate case, the HOMO (−0.221 eV) and HOMO−4 (−1.015 eV) consist mostly of the metal dz2 and dxz orbitals, respectively, while the HOMO−1 (−0.693 eV) primarily consists of ligand centered orbitals that has mainly p-orbital character (Figure 4A). The HOMO and HOMO−1 can be interpreted as the out-of-phase and in-phase combination of the dz2 and ligand-based orbitals. Upon bending, the distortion of geometry allows dxz to interact with the ligand's p orbital to form the HOMO (0.635 eV) and HOMO−5 (−1.797 eV). This new orbital interaction promotes the dxz to become the new HOMO at the bent geometry. In the heterocuprate, the same logic is applied to track the change of d-orbital energy during the structural distortion (Figure 4B). The HOMO (−1.300 eV) and HOMO−5 (−2.455 eV) consist of dz2 character and HOMO−1 (−2.145 eV) consists of dxz character. The bending motion allows the metal dxz and ligand orbital to form in-phase and out-of-phase combinations, HOMO−5 (−2.894 eV) and HOMO (−0.705 eV), respectively. Note that at the bent structure of the heterocuprate, the HOMO is much lower in energy compared to the bent homocuprate. This is expected, considering that the low-lying orbital of the cyano group lowers the ligand orbital energy (−2.455 eV). A detailed analysis is provided in the Supporting Information.

Walsh diagram of model system for homocuprate (A) and heterocuprate (B). Isodensity=0.05 a.u. and energies are given in eV.
Conclusion
In summary, we have developed a copper-catalyzed regioselective and stereospecific alkylation of unbiased internal allylic carbonates with a broad range of functionalized Grignard reagents. Modification of the ligand alters the reaction mechanism, which gives rise to different regioselectivities for the unbiased allylic systems. DFT calculations revealed that the origin of different mechanisms for the homo- and heterocuprate catalysts is derived from the different preferences for π-complex formation. Orbital analysis showed that the difference between the cyano- and alkyl ligands causes different preferences for catalyst bending, which is necessary for π-complexation. We anticipate our mechanism-based study of regioselective allylic alkylations will provide a deeper understanding of this class of reactions, which will lead to future advances in the selective functionalization of unbiased allylic systems.
Acknowledgments
Financial support was provided to U.K.T by W. W. Caruth, Jr. Endowed Scholarship, Welch Foundation (I-1748), National Institutes of Health (R01GM102604), American Chemical Society Petroleum Research Fund (59177-ND1), Teva Pharmaceuticals Marc A. Goshko Memorial Grant (60011-TEV), and Sloan Research Fellowship. The authors also thank our diverse collection of lab members for creating an environment that supported the success of this project. We thank the Institute for Basic Science in Korea for financial support (IBS-R010-A1). We thank Mr. Bohyun Park, Sangho So, Joon Heo and Jun-Hyeong Kim and Ms. Mina Son for fruitful discussions.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.

