

Kupplung von Reformatsky-Reagenzien und Arylchloriden ermöglicht durch Ylid-funktionalisierte Phosphanliganden
Professor Pierre Dixneuf zum 80. Geburtstag gewidmet
Abstract
Die Kupplung von Arylchloriden mit Reformatsky-Reagenzien ist eine erfolgversprechende, aber auch herausfordernde Strategie für die Synthese von α-Arylestern. So schränken diverse Nebenreaktionen ihre Anwendungsbreite erheblich ein. Die Limitierungen wurden nun durch die Anpassung von Ylid-funktionalisierten Phosphanen speziell auf die Anforderungen solcher Negishi-Kupplungen überwunden. Durch Verwendung eines Cyclohexyl-YPhos-Liganden, der einen ortho-Tolyl-Substituenten am Ylid-Grundgerüst trägt, wurden Rekordaktivitäten bei Palladium-katalysierten Arylierungen von Organozink-Reagenzien mit Arylelektrophilen erzielt. Dieser sehr elektronenreiche, sterisch anspruchsvolle Ligand ermöglicht die Verwendung von Arylchloriden in Kupplungen mit Reformatsky-Reagenzien bei Raumtemperatur. Die Anwendungsbreite umfasst vielfältig funktionalisierte Arylessig- und Arylpropionsäurederivate. Arylbromide und -chloride können selektiv gegenüber Triflat-Elektrophilen umgesetzt werden, was konsekutive Kupplungsstrategien ermöglicht.
Einleitung
Die α-Arylester-Einheit ist eine Schlüsselfunktionalität in biologisch aktiven Verbindungen und Pharmazeutika, darunter die nichtsteroidalen Antirheumatika Flurbiprofen, Ibuprofen, Naproxen und Pranoprofen sowie das Antihistaminikum Fexofenadin (Abbildung 1).1 Darüber hinaus sind α-Arylester und -amide wertvolle Synthone auf dem Weg zu Arylalkoholen, -aminen oder -nitrilen. Für die Synthese von α-Arylestern stehen mehrere Methoden zur Verfügung, die jedoch alle ihre individuellen Limitierungen haben (Schema 1).

Beispiele biologisch aktiver α-Arylalkylcarboxylate.

Synthesemöglichkeiten für α-Arylalkylcarboxylate.
Unkatalysierte C-C-Bindungsknüpfungen zwischen Arylelektrophilen und Enolaten, z. B. photochemische Reaktionen,2 Additionen an Arine,3 Reaktionen mit Arylbismut- oder Arylblei-Reagenzien4 sowie nukleophile aromatische Substitutionen an elektronenarmen Arenen,5 sind limitiert durch eine begrenzte Anwendungsbreite, toxische Reagenzien und/oder harsche Reaktionsbedingungen.
Friedel-Crafts-Alkylierungen von Arenen mit α-Halogencarbonylverbindungen ergeben ein Gemisch von Regioisomeren,6 während Reaktionen zwischen Arylboroxinen und Diazoestern durch die mehrstufige Synthese der Ausgangsstoffe eingeschränkt sind.7 Katalytische Carbonylierungen sind im großen Maßstab vorteilhaft, im Labormaßstab jedoch experimentell schwierig.8 Kupplungsreaktionen von Phenylboronsäuren oder Grignard-Reagenzien9 sind praktisch, aber durch die Verfügbarkeit des Arylnukleophils eingeschränkt.
In den letzten Jahrzehnten haben sich übergangsmetallkatalysierte α-Arylierungen von Carbonylverbindungen als ein wirksames Instrument für die Einführung von α-Arylesterfunktionalitäten als späte Synthesestufen entwickelt.1a, 5 Diese Reaktionen erfordern jedoch stark basische Bedingungen,10 und die erhöhte Acidität des arylierten Produkts im Vergleich zum Ausgangsmaterial führt oft zu unerwünschten Diarylierungen. In diesem Zusammenhang war die Verwendung von leicht basischen Zinkenolaten (Reformatsky-Reagenzien)11 ein großer Fortschritt. Diese Reagenzien sind aus α-Halogencarbonylverbindungen und Zink bzw. Zinkreagenzien oder direkt aus Carbonylverbindungen über Deprotonierungs-/Transmetallierungsstrategien leicht zugänglich.12 Hartwig et al. etablierten Pd-katalysierte Negishi-Kupplungen13 von Arylbromiden mit Reformatsky-Reagenzien als ein allgemein anwendbares Synthesekonzept für die Herstellung funktionalisierter α-Arylalkylester und -amide (Schema 2).14

Pd-katalysierte Negishi-Kupplung mit Reformatsky-Reagenzien.
Der Erfolg dieser Umsetzung hängt entscheidend von der Verwendung sterisch extrem anspruchsvoller, elektronenreicher Q-Phos-Liganden ab. Doch selbst mit diesem hochentwickelten Ligandensystem ist die Reaktion meist auf Arylbromide als Substrate beschränkt. Nur eine geringe Auswahl aktivierter Arylchloride wurde bei erhöhten Temperaturen erfolgreich umgesetzt, was die Autoren zu der folgenden Schlussfolgerung veranlasste “… studies are required to address the scope of the coupling (…) with chlororarenes” [15] Ähnliche Substrateinschränkungen sind auch bei metallaphotoredoxkatalysierten Kreuzkupplungen von Arylelektrophilen mit Halogencarboxylaten zu beobachten.16
Die Kupplung von Reformatsky-Reagenzien mit Arylchloriden17 stellt nach wie vor eine erhebliche Herausforderung dar, die selbst die hochentwickeltsten Katalysatorsysteme an ihre Leistungsgrenze bringt.15 Dies ist auf mehrere zusätzliche Herausforderungen zurückzuführen: 1) C- und O-metallierte Zinkenolate liegen im Gleichgewicht vor,12a und eine konkurrierende C-O-Bindungsbildung muss vermieden werden. 2) Das Arylierungsprodukt ist saurer als das Ausgangsmaterial, und Protonierungsgleichgewichte könnten zu diarylierten Nebenprodukten führen. 3) Die unkatalysierte Selbstkondensation von Esterenolaten verläuft schnell,18 sodass die oxidative Addition des Arylelektrophils noch schneller erfolgen muss, um mit dieser Hintergrundreaktion zu konkurrieren.19
Kürzlich haben wir Ylid-funktionalisierte Monophosphan-Liganden (YPhos) als Steuerliganden in der Katalyse etabliert.20 Palladium-YPhos-Komplexe lassen sich leicht aus Phosphoniumsalzen generieren, die in großer struktureller Vielfalt zugänglich sind. Die durch die Ylidgruppe induzierte hohe Donorstärke in Kombination mit sterisch anspruchsvollen Substituenten am Phosphor ermöglicht die Synthese von Pd-Katalysatoren mit außergewöhnlicher katalytischer Aktivität bei Buchwald-Hartwig-Aminierungen,21 Keton-Arylierungen22 und Kreuzkupplungen von Organolithium- und Grignard-Verbindungen.23 Die sperrigen Substituenten am Liganden erleichtern die Bildung hochreaktiver einfach-Ligand-gebundener Pd-Komplexe, die oxidative Additionen von desaktivierten Arylchloriden bei niedrigen Temperaturen ermöglichen.21a Diese Eigenschaften machten uns zuversichtlich, dass YPhos-Liganden bei der Kupplung von Zinkenolaten weit effektiver sein sollten als etablierte Ligandensysteme.
Ergebnisse und Diskussion
Um das Potenzial von YPhos-Pd-Komplexen als Katalysatoren in Negishi-Kupplungen zu testen, wählten wir die Kupplung des Reformatsky-Reagenzes 2 a mit 3-Chloranisol (1 a) als Testreaktion (Tabelle 1).
Nr. |
X |
Ligand L |
Additiv |
t [h] |
3 [%] |
|---|---|---|---|---|---|
1 |
Cl |
Q-Phos |
– |
12 |
4 |
2 |
Cl |
XPhos |
– |
12 |
16 |
3 |
Cl |
tBu-XPhos |
– |
12 |
18 |
4 |
Cl |
L1 |
– |
12 |
Spuren |
5 |
Cl |
L2 |
– |
12 |
35 |
6 |
Cl |
L3 |
– |
12 |
55 |
7 |
Cl |
L4 |
– |
12 |
30 |
8 |
Cl |
L5 |
– |
12 |
10 |
9 |
Cl |
L6 |
– |
12 |
16 |
10 |
Cl |
L3 |
– |
16 |
65 |
11 |
Cl |
L3 |
LiCl |
16 |
80 |
12 |
Cl |
L3 |
LiBr |
16 |
84 |
13 |
Cl |
L3 |
Et3N |
16 |
62 |
14 |
Cl |
L3 |
TMEDA |
16 |
97 |
15 |
Cl |
L3 |
DMPU |
16 |
82 |
16[b] |
Cl |
L3 |
TMEDA |
16 |
95 |
17[b,c] |
Cl |
L3 |
TMEDA |
16 |
96 (95)[d] |
18[b,c] |
Br |
L3 |
TMEDA |
16 |
97 |
19[b,c] |
I |
L3 |
TMEDA |
16 |
90 |
20[b,c] |
OTf |
L3 |
TMEDA |
16 |
68 |
- [a] Bedingungen: 0.25 mmol 1 a, 0.375 mmol 2 a, 2 mol % Pd2dba3, 4 mol % L, 1.5 Äquiv. Additiv, 0.3 mL THF, RT, 16 h. [b] 2 Äquiv. Additiv. [c] 1 mol % Pd2dba3. [d] Ausbeute an isoliertem Produkt basierend auf 0.5-mmol-Ansatz. Ausbeuten wurden mittels GC unter Verwendung von n-Tetradecan als internem Standard bestimmt.
Die Referenz auf diesem Gebiet, ein Q-Phos-Pd-System, erwies sich bei Raumtemperatur als inaktiv. Andere Hochleistungsliganden vom Buchwald- oder Fu-Typ sowie CataCXium A (Ad2PnBu) lieferten unbefriedigende Ausbeuten; Standard-Alkyl- oder -Arylphosphane waren ineffektiv (Tabelle 1, Nr. 1–3 und Tabelle S1). Bei der Verwendung von definierten Pd-Komplexen wurde keine signifikante Verbesserung beobachtet. Auch der einfache YPhos-Ligand L1 mit einer Methylgruppe am Ylid-Rückgrat wurde ohne Erfolg getestet. Die Einführung eines Arylsubstituenten am Ylid-Kohlenstoffatom (L2) führte jedoch zur gewünschten Reaktivität. Die Feinabstimmung des Ligandendesigns zeigte eine hohe Empfindlichkeit der Katalysatorleistung gegenüber der Ligandenstruktur, wobei sich die ortho-Tolylgruppe als optimaler Substituent erwies. Eine weitere Erhöhung des sterischen Anspruchs und eine Verringerung der Flexibilität der Ligandenstruktur durch Einführung einer Mesitylgruppe (L4) verringerte die katalytische Aktivität. Auch der elektronenreichere PtBu2-Ligand (L6) führte zu geringeren Ausbeuten, was darauf schließen lässt, dass die oxidative Addition nicht der limitierende Schritt im katalytischen Zyklus ist. Basierend auf diesen Erkenntnissen testeten wir auch den neu entworfenen Liganden L5 mit einem ortho-Anisyl-Substituenten (siehe SI), von dem wir annahmen, dass er die Transmetallierung durch Präkoordination des Zinkreagenzes erleichtern würde. Aber auch dieser Ligand führte zu keiner weiteren Verbesserung, was die hohen Anforderungen dieser Reaktion an das Ligandendesign unterstreicht.
Nachdem wir die beste Ligandenstruktur identifiziert hatten, richteten wir unsere Aufmerksamkeit auf eine weitere Verbesserung der Reaktionsbedingungen. Es ist bekannt, dass die Zugabe von Lösungsmitteln und Additiven die Lösungsstruktur von Zinkreagenzien verändert und dadurch deren Reaktivität und Stabilität beeinflusst.24 Es wurde berichtet, dass das Zinkenolat stabile Dimere bildet, was ein Grund für seine geringe Reaktivität sein könnte.24a, 24b Daher setzten wir TMEDA als Additiv ein, da bekannt ist, dass es die Bildung von einkernigen metallorganischen Spezies erleichtert. Zu unserer Freude erhöhte dieses Additiv die Effizienz der Reaktion stark, was wir auf eine höhere Konzentration von einkernigen Zinkspezies zurückführen.25 Aus diesem Grund testeten wir verschiedene Lösungsmittel, Metallhalogenide und organische Donormoleküle (Tabelle S2). THF erwies sich als das effektivste Lösungsmittel, TMEDA als das beste Additiv, das auch eine Reduzierung der Katalysatorbeladung auf 1 mol % ermöglichte.
Unter optimierten Bedingungen lieferte die Reaktion innerhalb von 16 Stunden bei Raumtemperatur nahezu quantitative Ausbeuten. Hohe Ausbeuten wurden auch ausgehend von den entsprechenden Arylbromiden (97 %),-iodiden (90 %) sowie -triflaten (68 %) erzielt. Der Katalysator begünstigt auch effektiv die Kupplung von Arylchloriden mit Standard-Zinkreagenzien wie Diethylzink, wobei das gewünschte Produkt 4 in 85 % Ausbeute erhalten wurde (Schema 3).

Pd-katalysierte Negishi-Kupplung mit Diethylzink.
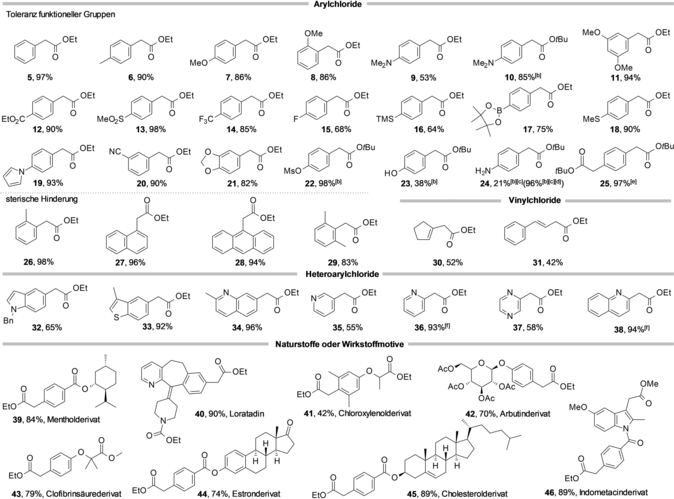
Eine Vielzahl von Aryl- und Heteroaryl- sowie Vinylchloriden wurde erfolgreich mit dem Reformatsky-Reagenz 2 a in guten Ausbeuten gekuppelt (Tabelle 2). Geeignete Substrate reichen von elektronenarmen (Pyrazinyl) bis zu sehr elektronenreichen (p-N,N-Dimethylaminophenyl) Aryl- oder Heteroarylchloriden, wobei die Anwendungsbreite sowohl sterisch sehr anspruchsvolle Gruppen (o,o-Dimethylphenyl) als auch koordinierende Pyridin- oder Thiophen-Heterocyclen umfasst. Eine Fülle von funktionellen Gruppen wird toleriert, darunter Trifluormethyl, Fluor, Ester, Trimethylsilyl, Nitril, Mesylat und sogar Pinakolboronat (Tabelle 2, 14–17, 20, 22). Die Effizienz der Kupplung ist für sterisch anspruchsvolle tert-Butylester-Substrate höher als für Ethylester-Substrate, sodass deren Verwendung sich für besonders anspruchsvolle Arylchloride empfiehlt. Die Leistungsgrenze des Systems wird für 4-Chlorphenol erreicht, das wahrscheinlich durch das Enolat deprotoniert wird, was zu einem extrem elektronenreichen Chlorphenolat führt, das sich der oxidativen Addition widersetzt. Dennoch konnte 4-Chlorphenol in einer Ausbeute von 38 % zu Verbindung 23 gekuppelt werden. Die Kupplung von 4-Chloranilin lieferte nur unbefriedigende Ergebnisse, aber 4-Bromanilin ergab Verbindung 24 in nahezu quantitativer Ausbeute trotz der freien NH-Gruppe (Tabelle 2).

|
- [a] Bedingungen: 0.5 mmol 1, 0.75 mmol 2 a oder 2 c, 1 mol % Pd2dba3, 2 mol % L3, 2 Äquiv. TMEDA, 0.3 mL THF, RT, 16 h, Ausbeuten an isoliertem Produkt. [b] 0.5 mol % Pd2dba3 und 1 mol % L3. [c] 2.2 Äquiv. 2 c. [d] Arylbromid wurde anstelle von Arylchlorid eingesetzt. [e] 3 Äquiv. 2 c. [f] Nach 40 h.
Die Toleranz gegenüber diesen funktionellen Gruppen ist bemerkenswert, insbesondere, wenn man bedenkt, dass Pd-YPhos-Systeme auch leistungsstarke Katalysatoren für C-N-Kreuzkupplungsreaktionen sind. Das entwickelte Reaktionsprotokoll ermöglicht auch die erfolgreiche Kupplung komplexerer Strukturen und zeigt damit sein Potenzial in der Endfunktionalisierung.4, 9d, 10g, 26 So konnten beispielsweise Derivate von Menthol, Cholesterol, Loratadin oder Clofibrinsäure sowie Estron und Arbutin in guten bis hohen Ausbeuten umgesetzt werden (Tabelle 2, 39–46).
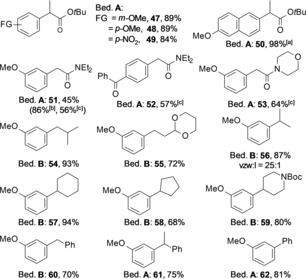
Als nächstes untersuchten wir die Anwendungsbreite der Transformation im Hinblick auf die Zinkreagenzien (Tabelle 3). Nicht nur lineare, sondern auch verzweigte Zinkenolate 2 d wurden erfolgreich gekuppelt, was angesichts der Bedeutung der Phenylpropenyl-Substruktur von erheblichem Interesse ist. Die Kupplung von α,α-disubstituierten Reformatsky-Reagenzien ergab zum jetzigen Stand nur unbefriedigende Ausbeuten (siehe SI). Ausgehend von kommerziell verfügbarem 2-Brom-6-methoxynaphthalin wurde der Naproxenester 50 in nahezu quantitativer Ausbeute erhalten. Amid-Enolate, auch mit Weinreb-typischer Reaktivität, die leicht aus Zn(TMP)2 oder Lithiumenolaten hergestellt werden können,14b, 27 wurden ebenfalls erfolgreich gekuppelt (Tabelle 3, 51–53). Der YPhos-Pd-Katalysator ermöglicht auch die Negishi-Kreuzkupplung28 von primären oder sekundären Alkyl-, Benzyl- und Arylzink-Reagenzien bei Raumtemperatur. Die besten Ergebnisse wurden bei Verwendung von Pd[COD]Cl2 anstelle von Pd2dba3 als Pd-Quelle erzielt (Bedingungen B). Zu diesem Zeitpunkt haben wir keine Erklärung dafür, warum Pd[COD]Cl2, das als Pd-Quelle bei der Kupplung von Zinkenolaten fast unwirksam ist, ein überlegener Pd-Vorläufer für andere Organozink-Reagenzien ist. Erfreulicherweise wurde fast keine Umlagerung zu linearen Produkten beobachtet. Das sekundäre Alkylzinkreagenz 2 i wurde mit einer Selektivität von 25:1 zugunsten des verzweigten Isomers umgesetzt (56); für das Benzylreagenz 2 n wurde ausschließlich das verzweigte Produkt beobachtet (61).

|
- Bedingung A: 0.5 mmol 1, 0.75 mmol 2, 1 mol % Pd2dba3, 2 mol % L3, 0.3 mL THF, 2 Äquiv. TMEDA, RT, 16 h, Ausbeuten an isoliertem Produkt. Bedingung B: 0.5 mmol 1, 0.75 mmol 2, 3 mol % Pd[COD]Cl2, 3 mol % L3, 0.3 mL THF, RT, 16 h, Ausbeuten an isoliertem Produkt. [a] Arylbromid wurde anstelle von Arylchlorid eingesetzt. [b] (Et2NCOCH2)2Zn, 2 mol % Pd2dba3, 4 mol % L3 und 1.5 Äquiv. LiBr. [c] Zinkreagenz wurde über das Lithiumenolat mit Zinkchlorid hergestellt, 0.5 mmol 1, 0.6 mmol Amid 2, 1 mol % Pd2dba3, 2 mol % L3, 1 mL THF.
Da vorläufige Untersuchungen (Tabelle 1) darauf hinwiesen, dass der YPhos-Pd-Komplex auch Bromide und Triflate zu den gewünschten Kupplungsprodukten umsetzt, untersuchten wir als nächstes eine mögliche Differenzierung zwischen den verschiedenen Elektrophilen. Zu diesem Zweck führten wir Konkurrenzexperimente durch, bei denen zwei Elektrophile gleichzeitig mit Zinkreagenzien behandelt wurden (Schema 4, Tabellen S3–S12).

Selektivität von Br/Cl-, Br/OTf- und Cl/OTf-Kuplungsreaktionen mit verschiedenen Zinkreagenzien. Bedingungen: 0.25 mmol (1 Äquiv.) Arylelektrophile, 1.1–1.5 Äquiv. Zinkreagenz 2, 1 mol % Pd2dba3, 2 mol % L3, 2 Äquiv. Additiv, 0.3 mL THF, RT oder 0 °C, 16 h. Ausbeuten wurden mittels GC unter Verwendung von n-Tetradecan oder Methyldecanoat als internem Standard bestimmt. Für detaillierte Bedingungen siehe SI Tabelle S3–S12.
Enolate, Alkyl- und Arylzinkreagenzien ergaben alle exzellente Selektivitäten zugunsten von Arylbromiden gegenüber Triflaten. Ein ähnliches Selektivitätsmuster wurde auch für Q-Phos beobachtet, während X-Phos deutlich weniger selektiv war (siehe Tabelle S13). Bemerkenswerterweise wurden sogar Arylchloride trotz ihrer geringeren inhärenten Abgangsgruppenfähigkeit bevorzugt gegenüber Triflaten gekuppelt.29 Dies deutet auf eine starke Präferenz der YPhos-Pd-Katalysatoren für weiche Nukleophile hin. Die 10:1-Selektivität von Arylbromiden gegenüber -chloriden liegt ebenfalls auf einem präparativ nützlichen Niveau.
Anhand der Synthese von Ibuprofen haben wir den synthetischen Nutzen des ausgeprägten Selektivitätsmusters des YPhos-Pd-Katalysators demonstriert. Die konsekutive Behandlung von p-Bromchlorbenzol oder p-Bromphenyltriflat mit Isobutylzink 2 g, anschließend mit Enolat 2 d in Gegenwart von YPhos-Pd, ergab das gewünschte Produkt 69 in hoher Selektivität und guter Ausbeute (Schema 5).

Synthese von Ibuprofen. Bedingung A: 1.3 Äquiv. 2 g, 1 mol % Pd2dba3, 2 mol % L3, 2 Äquiv. ZnBr2, 0.3 mL THF, RT, 16 h. Bedingung B: 1.3 Äquiv. 2 d, 1 mol % Pd2dba3, 2 mol % L3, 0.3 mL THF, 2 Äquiv. TMEDA, RT, 16 h. Hydrolyse: 5 Äquiv. TFA, 5.5 mL DCM, RT, 12 h.
Zusammenfassung
Zusammenfassend wurde gezeigt, dass ein sterisch anspruchsvoller YPhos-Pd-Katalysator bei der Negishi-Kreuzkupplung von Arylchloriden hocheffizient ist. Bereits die erste Katalysatorgeneration geht weit über den Stand der Technik bei der anspruchsvollen Kupplung von Reformatsky-Reagenzien hinaus. Das Screening einer Reihe verschiedener Phosphane zeigte, dass die Ligandenstruktur einen entscheidenden Einfluss auf die Katalysatoreffizienz hat, was zu drastischen Veränderungen, selbst bei scheinbar kleinen Variationen im Ligandendesign, führt. Das entwickelte Reaktionsprotokoll zeigt eine hohe Toleranz gegenüber einer Vielzahl von funktionellen Gruppen, was die Kupplung eines großen Substratspektrums einschließlich der Funktionalisierung komplexer, molekularer Strukturen ermöglichte. Darüber hinaus wurden hohe Selektivitäten mit sekundären Alkylzink-Reagenzien sowie für die Differenzierung zwischen Chlor-, Brom- und Triflat-Elektrophilen erreicht, was die konsekutive Funktionalisierung mit verschiedenen Nukleophilen ermöglicht. Insgesamt verbessert das vorgestellte Protokoll die Anwendungsmöglichkeiten von Zinkreagenzien in palladiumkatalysierten Kupplungsreaktionen erheblich und eröffnet neue Perspektiven für die Nutzung von Negishi-Kupplungen bei der Funktionalisierung komplexer Strukturen.
Experimentelles
Für die Kupplung von Zinkenolaten oder Aryl-Zink-Reagenzien wurde ein trockenes 20-mL-Reaktionsgefäß mit teflonbeschichtetem Rührkern mit Pd2dba3 (5.00 mg, 1 mol %, 21 Gew.-% Pd), L3 (5.80 mg, 2 mol %) befüllt und mit einer Aluminium-Bördelkappe mit teflonbeschichtetem Butylgummi-Septum verschlossen. Unter Ausschluss von Luft und Feuchtigkeit wurde THF (0.3 mL) zugegeben und das resultierende Reaktionsgemisch 1 h bei Raumtemperatur gerührt. Das Arylchlorid 1 (0.50 mmol) und Zinkreagenz 2 (0.75 mmol, 1.5 Äquiv.) sowie TMEDA (117 mg, 1.00 mmol, 2 Äquiv.) wurden über eine Spritze unter Argonatmosphäre zugegeben, und die resultierende Mischung wurde 16 h bei Raumtemperatur gerührt.
Für die Kupplung mit Alkylzink-Reagenzien wurde das trockene 20-mL-Reaktionsgefäß mit Pd[COD]Cl2 (4.33 mg, 3 mol %) und L3 (8.71 mg, 3 mol %) gefüllt und mit einer Aluminium-Bördelkappe mit teflonbeschichtetem Butylgummi-Septum verschlossen. Unter Ausschluss von Luft und Feuchtigkeit wurde THF (0.3 mL) zugegeben und das resultierende Reaktionsgemisch 1 h bei Raumtemperatur gerührt. Das Arylchlorid 1 a (0.50 mmol) und Zinkreagenz 2 (0.75 mmol, 1.5 Äquiv.) wurden über eine Spritze unter Argonatmosphäre zugegeben. Die resultierende Mischung wurde bei Raumtemperatur für 16 h gerührt.
Nach Abschluss der Reaktionen wurden diese mit Wasser (20 mL) gequencht und mit Et2O (3×20 mL) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter Kochsalzlösung (20 mL) gewaschen, über MgSO4 getrocknet und filtriert, und das Lösungsmittel wurde im Vakuum entfernt. Der Rückstand wurde mittels Säulenchromatographie (SiO2, Pentan/Et2O oder Cy/EtOAc) gereinigt.
Acknowledgements
Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) im Rahmen der Exzellenzstrategie – EXC-2033–390677874 – RESOLV und SFB TRR88 “3MET” sowie durch den Europäischen Forschungsrat (Starting Grant: YlideLigands 677749). Wir danken UMICORE AG & Co. KG für die zur Verfügung gestellten Katalysatoren, dem BMBF und dem Land NRW (Zentrum für molekulare Spektroskopie und Simulation solvensgesteuerter Prozesse “ZEMOS”) sowie dem CSC (Stipendium an Z. H.) und der Alexander von Humboldt-Stiftung (Stipendium an X.-J. W.) für finanzielle Unterstützung und M. Wüstefeld für HRMS-Messungen. Open Access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.
Conflict of interest
Die Autoren haben das Patent WO2019030 304 in Zusammenarbeit mit UMICORE AG & Co. KG, das YPhos-Liganden und -Komplexe abdeckt, angemeldet.

