

Bismutamide als einfache Vermittler hochselektiver Pn−Pn-Radikal-Kupplungsreaktionen (Pn=N, P, As)
Professor Siegfried Hünig zum 100. Geburtstag gewidmet
Abstract
Die kontrollierte Generierung wohldefinierter Radikalspezies und deren Nutzung in selektiven Folgereaktionen sind wichtige und herausfordernde Aufgaben in der Synthesechemie. Wir zeigen hier, dass einfache Bismutamidverbindungen des Typs [Bi(NAr2)3] in der Lage sind, bei Raumtemperatur in Lösung Aminylradikale (NAr2). freizusetzen. Als Produkte dieser Reaktionen werden die entsprechenden Hydrazine als Ergebnis hochselektiver N−N-Kupplungen erhalten. Die Anwendung dieser einfachen homolytischen Bi−Pn-Bindungsspaltung wurde auf die höheren Homologe der Pniktogene (Pn=N-As) erweitert: Homoleptische Bismutamide ermöglichen die hochselektive Dehydrokupplung von HPnR2 zu R2Pn−PnR2. NMR- und EPR-spektroskopische Untersuchungen sowie Einkristallröntgenstrukturanalyse und DFT-Rechnungen zeigen, dass geringe homolytische Bi−N-Bindungsspaltungsenergien vorliegen, dass Radikalkupplungen in der Koordinationssphäre des Bismutzentrums anzunehmen sind und dass diese Reaktionen über die Einstellung elektronischer und sterischer Parameter effektiv gesteuert werden können.
Das kontrollierte Freisetzen radikalischer Spezies und deren darauffolgende Verwendung in selektiven Bindungsknüpfungen ist noch immer eine außerordentliche Herausforderung der Synthesechemie.1 Jüngere Fortschritte in der Chemie der schweren p-Block-Elemente zeigen deren Potential für die Entwicklung kontrollierter Radikalreaktionen.2, 3 Beispiele reichen von CH-Aktivierungsreaktionen durch Zinnradikale,4 über die Aktivierung von P4 und S8 durch Bismutradikale,5 bis hin zu Bismut-katalysierten Radikal-Dehydrokupplungsreaktionen,6 Radikal-Cycloisomerisierungen von Iodoolefinen7 und kontrollierten, radikalischen Olefinpolymerisationen.8 Die letzten Entwicklungen setzen dabei vor allem auf isolierbare Radikalspezies9 und die homolytische Bindungsspaltung von E−E-Bindungen,3a, 3b wohingegen die radikalstarterfreie Homolyse von E−X-Bindung als Radikalquelle von X. nur wenig untersucht ist (E=schweres p-Block Element; X=C, N, O). So wurde beispielsweise die homolytische Bindungsspaltung von E−C-Bindungen in der kontrollierten, radikalischen Polymerisation von Olefinen genutzt, was jedoch die Zugabe eines kohlenstoffzentrierten Radikals als Starter zum Olefinmonomer benötigt.8 Die Homolyse von Bi−O-Bindungen als geschwindigkeitsbestimmender Schritt der industriell relevanten Ammoxidation von Propen zu Acrylnitril an einen Bi2O3⋅MoO3 Katalysator (SOHIO Prozess) wurde lange diskutiert.10
Aminylradikale, [NR2]., spielen eine wichtige Rolle in biologischen Prozessen11 und deren Anwendung in neu aufkommenden Syntheseprotokollen erfreut sich stetig steigendem Interesse.12 Allerdings ist deren Verwendung auf Grund ihrer hohen Reaktivität und dem Mangel an selektiven Methoden zur Generierung unter milden Bedingungen weiterhin begrenzt.12a Dementsprechend kann die Homolyse von E−N-Bindungen eine vielversprechende Perspektive für die Bildung von Aminylradikalen darstellen. Tatsächlich wurde die homolytische E−N-Bindungsspaltung schon vorgeschlagen, z. B. in der Synthese von monodispersen Nanopartikeln ausgehend von [Bi(N(SiMe3)2)3] oder in CH-Aktivierungs-/ C−C-Kupplungssequenzen ausgehend von [BiCl2(N(SiMe3)(Mes*))] (Mes*=2,4,6-tBu3C6H2).13 Zudem konnten Pyrazolylradikale durch Oxidation des Pyrazolidanions mit BiCl3 dargestellt werden.14 Allerdings konnte das Freisetzen von Aminylradikalen ausgehend von wohldefinierten Vorläufern durch experimentell nachgewiesene, homolytische Bi−N-Bindungsspaltung unter milden Reaktionsbedingungen und die darauffolgende Verwendung in selektiven Bindungsknüpfungsereignissen bis jetzt noch nicht beschrieben werden.
Wir zeigen hier, dass Bismutamide der Form [Bi(NAr2)3] bereitwillig Aminylradikale freisetzen, welche unter milden Reaktionsbedingungen hochselektive N−N-Bindungsknüpfungen ermöglichen. Die Strategie der einfachen Bi−Pn-Bindungshomolyse konnte auf die höheren Homologe (Pn=N, P, As) erweitert werden.
Eine begrenzte Anzahl homoleptischer Bismutamide mit Alkyl-15 oder Silylsubstituenten15b, 16 ist zur Zeit bekannt. Ebenfalls sind wenige Bismutverbindungen mit Amidliganden, welche eine Arylgruppe (NArR)− enthalten, beschrieben. Diese zeigen zum Teil ein ungewöhnliches Reaktionsverhalten, welches in vielen Fällen nicht erklärt werden konnte (R=H, Si(alkyl)3; Supp. Inf.). Im Gegensatz dazu ist lediglich ein einziges homoleptisches Bismutamid des Typs [Bi(NAr2)3] literaturbekannt, welches auf Grund seines einfachen Arylsubstituenten (Ar=Ph, 1-H) heraussticht.17, 18
Unser Ziel war die Synthese einer Serie homoleptischer Bismutdiarylamide, [BiN(Ar2)3] (1-X), mit einfachem Substitutionsmuster, welches keinen extremen sterischen Anspruch in die Verbindungen einbringt (Schema 1 a; X=H, Me, OMe, Br, Ph, CF3). Dafür war es nötig, unterschiedliche Salzeliminierungsprotokolle (Route A, B) sowie eine Transaminierungsreaktion (Route C) zu entwickeln, da größere Unterschiede der Löslichkeit, Reaktivität und Stabilität der Edukte, Produkte und Nebenprodukte auftraten (Supp. Inf.). Die Verbindungen 1-X wurden als orangene bis violette Feststoffe in moderaten (51 %, X=OMe) bis exzellenten (93 %, X=Ph) Ausbeuten erhalten. Die Strukturen von 1-H,17 1-Me, 1-Br und 1-Ph im Festkörper wurden mittels Einkristallröntgenstrukturanalyse bestimmt und zeigen in allen Fällen isostrukturelle Beziehungen (trikline Raumgruppe P ). Die Verbindungen zeigen typische Molekülstrukturen mit trigonal pyramidaler Koordinationsgeometrie um die zentralen Bismutatome (N−Bi−N, 94.2°–107.7°; siehe Schema 1 b für 1-Me und Supp. Inf. für Details). In 1-Me beträgt der intermolekulare Bi⋅⋅⋅Bi-Abstand 3.79 Å, was auf eine schwache Bi⋅⋅⋅Bi-Wechselwirkung im Festkörper hinweist (Supp. Inf.).19 Die Bi−N-Bindungslängen in 1-H, 1-Me und 1-Br sind praktisch unabhängig von der Natur des Substituenten X in para-Position der Arylfunktion (2.15–2.19 Å). Die leicht verlängerten Bi−N-Bindungen in 1-Ph (2.16–2.23 Å) wurden den intramolekularen π-Wechselwirkungen zwischen zwei [(N(C6H4Ph)2]− Liganden zugeschrieben. Die N-Atome sind nur schwach pyramidalisiert (∑C−N−C/Bi, 352°-360°). NMR-spektroskopische Analysen der Verbindungen 1-X zeigen Signalmuster, welche sechs magnetisch äquivalenten Arylgruppen ohne signifikante Rotationsbarrieren bei Raumtemperatur in Lösung entsprechen (Schema 2 b (oben) zeigt das 1H-NMR-Spektrum von 1-Me). Allerdings stellte sich schnell heraus, dass die Verbindungen 1-X nur eine begrenzte Lebenszeit in Lösung haben. Die Verbindungen durchlaufen bei 23 °C in Benzol eine selektive Reaktion, bei der sich auch ein schwarzer Feststoff bildet (die 1H-NMR-spektroskopische Verfolgung dieses Prozesses ist in Schema 2 b für 1-Me gezeigt).20 Als Produkte dieser Reaktion wurden die Hydrazine 3-X identifiziert, d. h. die Verbindungen 1-X dienen als Vorläufer für selektive N−N-Kupplungsreaktionen (Schema 2 a). Die hohen (bis quantitativen) Ausbeuten dieser Reaktionen zeigen, dass es sich bei dem schwarzen Feststoff, auf Grund der Atombilanz, hauptsächlich bzw. ausschließlich um elementares Bi0 handelt. Bei der Durchführung der Reaktion 1-Me → 3-Me unter Lichtausschluss wurde ein identisches Ergebnis erhalten, sodass es sich um einen thermisch induzierten Prozess handelt. Mittels EPR-spektroskopischer Untersuchung der Reaktion konnte im Fall von 1-Me das Aminylradikal [N(tol)2]. (2-Me) als Intermediat beobachtet werden (tol=4-Me-C6H4; Schema 2 c und Supp. Inf.). Für isoliertes 3-Me wurde unter gleichen Bedingungen (wie in der Literatur beschrieben) kein Signal im EPR-Spektrum beobachtet.20b Dies zeigt, dass die homolytische Bi−N-Bindungsspaltung der Ursprung des Aminylradikals ist. Dies ist nach unserem besten Wissen der erste experimentelle Beweis einer Bi−N-Homolyse bei milden Bedingungen in kondensierter Phase. Anhand unterschiedlicher Substituenten X in para-Position des Arylrückgrats der Verbindungen 1-X wurde der elektronische Einfluss auf die Eigenschaften dieser Verbindungsklasse untersucht. Mittels 1H-NMR-Spektroskopie wurden für jede Verbindung Halbwertszeiten bestimmt. Für die elektronenreichen Verbindungen 1-Me und 1-OMe zeigte sich ein rascher Reaktionsverlauf (t1/2=1.2–1.4 h). Im Gegensatz dazu wurde für die Verbindungen 1-H, 1-Br und 1-Ph, welche geringere Elektronendichten in den Phenylengruppen aufweisen, deutlich längere Reaktionszeiten beobachtet (t1/2=63–84 h). Dieser Trend wurde durch die elektronenärmste Verbindung 1-CF3 bestätigt, welche bei 23 °C in Lösung über Tage stabil ist. Bei der intermolekularen Reaktion von drei Äquivalenten HN(4-Me-C6H4)2 mit Bi(NMe2)3 (4) in Benzol waren HNMe2 und 3-Me die benzollöslichen Hauptprodukte (Supp. Inf.).
). Die Verbindungen zeigen typische Molekülstrukturen mit trigonal pyramidaler Koordinationsgeometrie um die zentralen Bismutatome (N−Bi−N, 94.2°–107.7°; siehe Schema 1 b für 1-Me und Supp. Inf. für Details). In 1-Me beträgt der intermolekulare Bi⋅⋅⋅Bi-Abstand 3.79 Å, was auf eine schwache Bi⋅⋅⋅Bi-Wechselwirkung im Festkörper hinweist (Supp. Inf.).19 Die Bi−N-Bindungslängen in 1-H, 1-Me und 1-Br sind praktisch unabhängig von der Natur des Substituenten X in para-Position der Arylfunktion (2.15–2.19 Å). Die leicht verlängerten Bi−N-Bindungen in 1-Ph (2.16–2.23 Å) wurden den intramolekularen π-Wechselwirkungen zwischen zwei [(N(C6H4Ph)2]− Liganden zugeschrieben. Die N-Atome sind nur schwach pyramidalisiert (∑C−N−C/Bi, 352°-360°). NMR-spektroskopische Analysen der Verbindungen 1-X zeigen Signalmuster, welche sechs magnetisch äquivalenten Arylgruppen ohne signifikante Rotationsbarrieren bei Raumtemperatur in Lösung entsprechen (Schema 2 b (oben) zeigt das 1H-NMR-Spektrum von 1-Me). Allerdings stellte sich schnell heraus, dass die Verbindungen 1-X nur eine begrenzte Lebenszeit in Lösung haben. Die Verbindungen durchlaufen bei 23 °C in Benzol eine selektive Reaktion, bei der sich auch ein schwarzer Feststoff bildet (die 1H-NMR-spektroskopische Verfolgung dieses Prozesses ist in Schema 2 b für 1-Me gezeigt).20 Als Produkte dieser Reaktion wurden die Hydrazine 3-X identifiziert, d. h. die Verbindungen 1-X dienen als Vorläufer für selektive N−N-Kupplungsreaktionen (Schema 2 a). Die hohen (bis quantitativen) Ausbeuten dieser Reaktionen zeigen, dass es sich bei dem schwarzen Feststoff, auf Grund der Atombilanz, hauptsächlich bzw. ausschließlich um elementares Bi0 handelt. Bei der Durchführung der Reaktion 1-Me → 3-Me unter Lichtausschluss wurde ein identisches Ergebnis erhalten, sodass es sich um einen thermisch induzierten Prozess handelt. Mittels EPR-spektroskopischer Untersuchung der Reaktion konnte im Fall von 1-Me das Aminylradikal [N(tol)2]. (2-Me) als Intermediat beobachtet werden (tol=4-Me-C6H4; Schema 2 c und Supp. Inf.). Für isoliertes 3-Me wurde unter gleichen Bedingungen (wie in der Literatur beschrieben) kein Signal im EPR-Spektrum beobachtet.20b Dies zeigt, dass die homolytische Bi−N-Bindungsspaltung der Ursprung des Aminylradikals ist. Dies ist nach unserem besten Wissen der erste experimentelle Beweis einer Bi−N-Homolyse bei milden Bedingungen in kondensierter Phase. Anhand unterschiedlicher Substituenten X in para-Position des Arylrückgrats der Verbindungen 1-X wurde der elektronische Einfluss auf die Eigenschaften dieser Verbindungsklasse untersucht. Mittels 1H-NMR-Spektroskopie wurden für jede Verbindung Halbwertszeiten bestimmt. Für die elektronenreichen Verbindungen 1-Me und 1-OMe zeigte sich ein rascher Reaktionsverlauf (t1/2=1.2–1.4 h). Im Gegensatz dazu wurde für die Verbindungen 1-H, 1-Br und 1-Ph, welche geringere Elektronendichten in den Phenylengruppen aufweisen, deutlich längere Reaktionszeiten beobachtet (t1/2=63–84 h). Dieser Trend wurde durch die elektronenärmste Verbindung 1-CF3 bestätigt, welche bei 23 °C in Lösung über Tage stabil ist. Bei der intermolekularen Reaktion von drei Äquivalenten HN(4-Me-C6H4)2 mit Bi(NMe2)3 (4) in Benzol waren HNMe2 und 3-Me die benzollöslichen Hauptprodukte (Supp. Inf.).

a) Synthese der Aryl-substituierten, homoleptischen Bismutamide: Route A: 1-H, 1-Me, 1-OMe; Route B: 1-OMe, 1-Br, 1-Ph; Route C: 1-CF3. b) Molekülstruktur von 1-Me im Festkörper. Ellipsoide repräsentieren eine Aufenthaltswahrscheinlichkeit von 50 %. Wasserstoffatome werden aus Gründen der Übersichtlichkeit nicht gezeigt. Ausgewählte Bindungslängen [Å] und -winkel [°]: Bi1−N1, 2.1598(19); Bi1−N2, 2.158(2); Bi1−N3, 2.185(2); N1−Bi1−N2, 100.68(8); N1−Bi1−N3, 97.92(8); N2−Bi1−N3, 95.03(8).

a) Selektive Bildung der Hydrazine 3-X aus den Bismutamiden (1-X) unter N−N-Radikalkupplung (Ausbeuten bestimmt mittels 1H-NMR-Spektroskopie). b) NMR-spektroskopische Beobachtung der Reaktion 1-Me → 3-Me (Sterne zeigen das Signal von C6D6). c) EPR-spektroskopische Verfolgung der Reaktion 1-Me → 3-Me mit Detektion von [N(tol)2]. (2-Me) (für Details siehe Supp. Inf.).
Diese neuen Erkenntnisse über Diarylamide, [Bi(NAr2)3] (1-X), erlauben nun erstmals ein ausführliches Verständnis der Eigenschaften und spektroskopischen Daten dieser Verbindungsklasse. Eine tiefergehende Untersuchung des bis zu diesem Zeitpunkt ersten Vertreters 1-H zeigten lediglich die vier erwarteten Signale im 13C-NMR-Spektrum in C6D6; im Gegensatz dazu waren für diese Verbindung in CD2Cl2 zuvor sieben Signale im 13C-NMR-Spektrum berichtet worden.17 Unsere Analysen zeigen, dass diese sich aus einer Produktmischung aus 1-H (4 Signale) und dem N−N-Kupplungsprodukt 3-H (4 Signale) ergeben, wobei zwei der Resonanzen überlappen. Durch eine optimierte Synthese konnte Verbindung 1-H im Multi-Gramm-Maßstab in 88 % Ausbeute analysenrein erhalten werden (verglichen mit einer ursprünglich berichteten Ausbeute von 30 %).
Anhand von DFT-Rechnungen wurden mechanistische Aspekte der Reaktion von 1-Me zu 3-Me näher untersucht (in folgender Diskussion ist R=4-Me-C6H4). Die homolytische Bi−N-Bindungsspaltung von 1-Me ist mit ΔΔG=36.8 kcal mol−1 deutlich gegenüber der heterolytischen Bi−N-Bindungsspaltung begünstigt. Umgekehrt ist die heterolytische Bi−N Bindungsspaltung beim protonierten, kationischen Derivat [Bi(NR2)2(HNR2)]+ bevorzugt (ΔΔG=24.1 kcal mol−1; für Details, siehe Supp. Inf.).
Die Ergebnisse der Energiedekompositionsanalyse der Bi−N-Bindung in 1-Me zeigten ähnliche stabilisierende Anteile der Orbital- (45 %) und elektrostatischen (46 %) Wechselwirkung zur Bi−N-Bindung, wohingegen Dispersionswechselwirkungen zwar signifikant (9 %), aber nicht ausschlaggebend zur Stabilität beitragen. Die Eliminierung von 3-Me aus 1-Me unter Bildung des Triplett-Bismutinidens 3[Bi(NR2)] konnte als erster Schritt der Reaktion 2 1-Me → 2 Bi0 + 3 3-Me auf Grund der hohen Gibbs-Energie (ΔG=46.3 kcal mol−1) ausgeschlossen werden (Schema 3). Die Bildung des korrespondierenden Singulett-Bismutinidens zeigt (wie auch Benzoladdukte desselben) eine noch höhere Gibbs-Energie (Supp. Inf.).3c Im Gegensatz dazu erscheint die Bildung eines Dibismutans entsprechend der Reaktion 2 1-Me → [Bi2(NR2)4] + 3-Me aus thermodynamischer Sicht realistischer (ΔG=12.7 kcal mol−1).21 Die zweistufige Eliminierung von zwei Äquivalenten 3-Me von [Bi2(NR2)4] erst zum Dibismuten [Bi2(NR2)2] (ΔG=36.4 kcal mol−1) und dann zu Bi2 (ΔG=21.4 kcal mol−1) ist energetisch nicht möglich. Stattdessen wird das Verhältnis Bi:NR2 wahrscheinlich durch die Bildung von Bismacyclen durch Reaktionen wie 2 [Bi2(NR2)4] → [Bi4(NR2)4] + 2 3-Me (ΔG=18.0 kcal mol−1) erhöht. Durch die Bildung von Bismutclustern22 als möglichen Zwischenstufen wird schlussendlich Bi0 erhalten. Unsere Berechnungen weisen darauf hin, dass die Bildung des Bismutmetalls eine wichtige Triebkraft dieser Reaktion ist (z. B.: [Bi4(NR2)4] → 4 Bi0(bulk) + 2 3-Me (ΔG=−62.1 kcal mol−1)). Die Nettoreaktion 2 1-Me → 2 Bi0(bulk) + 3 3-Me ist mit ΔG=−9.4 kcal mol−1 exergonisch. Wir gehen davon aus, dass die beschriebenen Einzelschritte durch Radikale wie [NR2]. katalysiert werden können (Supp Inf.). Die homolytische Bi−N-Bindungsspaltung von 1-Me (ΔG=25.4 kcal mol−1) oder von [Bi2(NR2)4] (ΔG=28.9 kcal mol−1) würde ausreichende Konzentrationen der Radikalspezies, wie auch mittels EPR-Spektroskopie beobachtet (vide supra), bereitstellen. Untersuchungen zur Kinetik der Bildung von 3-Me aus 1-Me unterstreichen die Komplexität dieser Reaktionssequenz und deuten auf eine Konzentrationsabhängigkeit des Mechanismus dieser Radikalreaktion hin (Supp. Inf.).

Mögliche Reaktionsschritte in der Bildung von 3-Me aus 1-Me (ungünstige [günstigere] Schritte sind in blau [schwarz] gezeigt; Energieniveaus sind auf ΔG skaliert); Werte für ΔH (Normalschrift) und ΔG (kursiv) sind in kcal mol−1 gegeben.
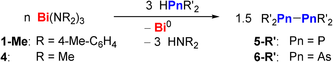
Um die Anwendung der Bismutverbindungen als Reagenzien für die selektive Homokupplung von leichteren Gruppe-15-Verbindungen zu erweitern, haben wir uns sekundären Phosphanen als möglichen Substraten zugewandt. Für die Dehydrokupplung von sekundären Phosphanen zu Diphosphanen, R2P−PR2 (wichtige Bausteine für die Synthese zweizähniger Liganden),23 wurde lange Zeit die Verwendung von Übergangsmetallkomplexen benötigt.24 Erst kürzlich wurden erste Berichte zur Durchführung dieser Reaktion mittels Hauptgruppenverbindungen veröffentlicht. Carbenoide, NHCs, Tri(aryl)borane, der Radikalstarter “Vazo 88” und KOtBu in Anwesenheit verschiedener Wasserstoffakzeptoren sind in der Lage Diphosphane aus sekundären Phosphanen zu generieren. Sie sind jedoch alle auf Substrate mit mindestens einer Arylgruppe limitiert.25, 26-28 Lediglich für die Organozinnverbindung Sn(C5Me5)2Cl2 wurde beschrieben, dass sie in der Lage ist HPCy2 als einziges Beispiel eines Dialkylphosphans einer Dehydrokupplung zu unterziehen (Cy=Cyclohexyl).29 Diese Reaktionen laufen sogar katalytisch ab (10 mol % Sn(C5Me5)2Cl2), erfordern aber sowohl lange Reaktionszeiten (3 Tage) und erhöhte Temperaturen (60 °C), als auch aufwendigere Durchführungen (regelmäßiges Entfernen von H2) und haben lediglich geringe Ausbeuten von 40 % Cy2P−PCy2. Berichte von schweren Hauptgruppenverbindungen L1Bi−PPh2 (postuliert) und L2Pb−PPh2 (isoliert), aus welchen das Kupplungsprodukt Ph2P−PPh2 (5-Ph) isoliert wurde, motivierten uns das Potential der Bismutamide in Dehydrokupplungsreaktionen von sekundären Phosphanen zu testen (L1/L2=sterisch anspruchsvoller mono-/dianionischer Ligand).5a, 30 Bei der Reaktion von 1-Me mit drei Äquivalenten HPPh2 bei 23 °C in Benzol wurde eine direkte und quantitative Bildung des Kupplungsproduktes 5-Ph (Tabelle 1, Eintrag 1) beobachtet. Zusätzlich fiel ein schwarzer Feststoff (wahrscheinlich Bi0) aus und einzig HN(4-Me-C6H4) war als benzollösliches Nebenprodukt zu beobachten. Die sterisch anspruchsvolleren Phosphane HP(Xyl)2 und HPMes2 konnten auch bei Raumtemperatur mit hohen bis quantitativen Ausbeuten gekoppelt werden, jedoch mussten zwei Äquivalente Bi(NMe2)3 (4) als Bismutamid eingesetzt werden, und beim zweiten Fall waren ebenfalls verlängerte Reaktionszeiten nötig (Einträge 2,3). Auch funktionelle Gruppen im Arylrückgrat werden toleriert und Diarylphosphane mit Elektronen-donierenden oder -ziehenden Substituenten (OMe, Cl, CF3) in para-Position wurden in guten Ausbeuten gekoppelt (Ausbeuten 92–99 %, Einträge 4–6). Auch die synthetisch anspruchsvollere Dehydrokupplung von Dialkylphosphanen war erfolgreich: Die Reaktion von 1-Me mit 3 Äquivalenten HPCy2 bei Raumtemperatur ergab 5-Cy in quantitativen Ausbeuten innerhalb von 1.5 h (Eintrag 7). Ähnlich verhielten sich HPiPr2 und HP(cyclo-pentyl)2 mit ebenfalls exzellenten Ausbeuten (Einträge 8,9). Generell werden Dehydrokupplungsreaktionen sekundärer Phosphane nicht nur durch Elektronen-donierende Substituenten am Phosphor erschwert, sondern auch durch höheren sterischen Anspruch am Phosphoratom.25b, 25d, 28, 29 Dementsprechend ist es bemerkenswert, dass nicht nur HPMes2 (Eintrag 3), sondern sogar HPtBu2 und HPAd2 mit ihren sterisch hochanspruchsvollen Alkylsubstituenten quantitativ in ihre Kupplungsprodukte 5-tBu und 5-Ad überführt werden können, wenn auch mit modifizierten experimentellen Bedingungen (Einträge 10,11). Die Reaktion des sekundären Arsans HAsPh2 mit 1-Me (oder 4) wurde als Grundsatznachweis durchgeführt, wobei das Kopplungsprodukt 6-Ph in 80 % Ausbeute selektiv erhalten wurde (Eintrag 12 und Supp. Inf.). Bei der Bildung von Ph2P−PPh2 wird in der Literatur die homolytische Spaltung von L1Bi−PPh2- und L2Pb−PPh2-Bindungen diskutiert (vide supra).5a, 30 Deshalb wurde die Reaktion von 4 mit HPMes2 mittels EPR Spektroskopie untersucht und ein schwaches Signal erhalten. Es wird vorgeschlagen, dieses Signal einem P-zentrierten Radikal (giso=2.007, a(31P)=270 MHz (96 G))31 zuzuordnen, was dementsprechend auf einen radikalischen Reaktionsschritt hinweist (für Details und Diskussion siehe Supp. Inf.). Im Gegensatz dazu wurden für die meisten von Hauptgruppenverbindungen vermittelten Phosphan-Dehydrokupplungen polare Reaktionsmechanismen diskutiert.25a-25c, 26, 28, 29 Auch für das einzige literaturbekannte Beispiel einer Hauptgruppenverbindung, welche die bis dato herausfordernde Dehydrokupplung eines Dialkylphosphans ermöglicht, wurden polare Reaktionspfade vorgeschlagen.26, 29 Katalytische Protokolle auf Basis von Übergangsmetallkomplexen für die Dehydrokupplung von Phosphanen benötigen erhöhte Temperaturen, längere Reaktionszeiten und zeigen Grenzen in der Substratbreite (siehe Supp. Inf.).24
# |
Reagenz |
Substrat |
n |
Bedingungen |
Ausbeute |
|---|---|---|---|---|---|
1 |
1-Me |
HPPh2 |
1 |
RT, <7 min |
>99 % |
2 |
1-Me |
HP(Xyl)2[a] |
2 |
RT, 3 h |
90 % |
3 |
4 |
HPMes2 |
2 |
RT, 16 h |
>99 % |
4 |
1-Me |
HP(4-OMe-C6H4)2 |
1.5 |
RT, <2 h |
92 % |
5 |
1-Me |
HP(4-Cl-C6H4)2 |
1.2 |
RT, <2 h |
97 % |
6 |
1-Me |
HP(4-CF3-C6H4)2 |
2 |
RT, <3 h |
>99 % |
7 |
1-Me |
HPCy2 |
1 |
RT, 1.5 h |
>99 % |
8 |
1-Me |
HPiPr2 |
1.2 |
RT, <6 h |
99 % |
9 |
4 |
HP(cyclo-pentyl)2 |
2 |
RT, <3 h |
94 % |
10 |
4 |
HPtBu2 |
2 |
60 °C, 1 d |
>99 % |
11 |
4 |
HPAd2[b] |
6 |
60 °C, 2 d |
>99 % |
12 |
1-Me |
HAsPh2 |
1 |
−78 °C, 0.5 h |
80 % |
- Ausbeuten bestimmt mittels 1H- und/oder 31P-NMR-Spektroskopie. [a] Xyl=3,5-Me2-C6H3. [b] Ad=adamantyl.
Zusammenfassend wurde das Bismutdiarylamid [Bi(NPh2)3] tiefergehend untersucht, was ein umfassenderes Verständnis dieser Verbindungsklasse ermöglichte. Bemerkenswerte Eigenschaften dieser Verbindungen sind die Freisetzung von Aminylradikalen unter milden Reaktionsbedingungen und die Vermittlung von hochselektiven N−N-Bindungsknüpfungen. Eine homolytische Bi−N-Bindungsspaltung wurde erstmals experimentell nachgewiesen. Sowohl der Einfluss der elektronischen Parameter auf die Freisetzung der Aminylradikale als auch fundamentalen Schritte des Reaktionsmechanismus wurden aufgezeigt. Einfache, homoleptische Bismutamide vermitteln zudem hochselektive Dehydrokupplungsreaktionen von HPnR2 zu R2Pn−PnR2 (Pn=N-As). Diese Ergebnisse zeigen das Potential von Bismutverbindungen für den Einsatz zur kontrollierten Generierung und darauffolgende synthetische Anwendung von Radikalen wie [PnR2].. Die Fähigkeit des Bismut Radikale freizusetzen und gleichzeitig, auf Grund des großen Atomradius, sterisch anspruchsvolle Liganden aufzunehmen, grenzt bismutbasierte Methoden von anderen literaturbekannten Methoden in diesem Forschungsfeld ab.
Conflict of interest
Die Autoren erklären, dass keine Interessenkonflikte vorliegen.

