

Prominin-1 (CD133) modulates the architecture and dynamics of microvilli
Abstract
Prominin-1 is a cell surface biomarker that allows the identification of stem and cancer stem cells from different organs. It is also expressed in several differentiated epithelial and non-epithelial cells. Irrespective of the cell type, prominin-1 is associated with plasma membrane protrusions. Here, we investigate its impact on the architecture of membrane protrusions using microvilli of Madin-Darby canine kidney cells as the main model. Our high-resolution analysis revealed that upon the overexpression of prominin-1 the number of microvilli and clusters of them increased. Microvilli with branched and/or knob-like morphologies were observed and stimulated by mutations in the ganglioside-binding site of prominin-1. The altered phenotypes were caused by the interaction of prominin-1 with phosphoinositide 3-kinase and Arp2/3 complex. Mutation of tyrosine 828 of prominin-1 impaired its phosphorylation and thereby inhibited the aforementioned interactions abolishing altered microvilli. This suggests that the interplay of prominin-1-ganglioside membrane complexes, phosphoinositide 3-kinase and cytoskeleton components regulates microvillar architecture. Lastly, the expression of prominin-1 and its mutants modified the structure of filopodia emerging from fibroblast-like cells and silencing human prominin-1 in primary hematopoietic stem cells resulted in the loss of uropod-associated microvilli. Altogether, these findings strengthen the role of prominin-1 as an organizer of cellular protrusions.
1 INTRODUCTION
Prominin-1 (alias CD133) is the first member of an evolutionarily conserved membrane glycoprotein family. It is broadly expressed among various tissues, particularly in differentiated epithelia, glial and photoreceptor cells.1-7 Besides, several organ-specific somatic stem cells, notably those in the neural and hematopoietic systems, were identified and isolated based on its expression.7, 8 Prominin-1 also label facultative and cancer stem cells.9-11
Prominin-1 is selectively enriched in protruding subdomains of the plasma membrane, such as microvilli and the primary cilium at the apical surface of epithelial cells.7, 12, 13 It is also concentrated in stereocilia and motile cilia of the epididymis and endometrium, respectively.3, 4 The specific subcellular localization of prominin-1 is likewise observed in non-epithelial cells, such as hematopoietic stem cells (HSCs). 1, 5, 14 Upon transfection of Chinese hamster ovary (CHO) cells, prominin-1 is strongly enriched in filopodia.7 Furthermore, prominin-1 is released into various body fluids in association with small membrane vesicles.15-18 They originate from the tips of microvilli and cilia, where prominin-1+ budding structures were detected.13, 19 These observations highlight the membrane dynamics of cellular protrusions.
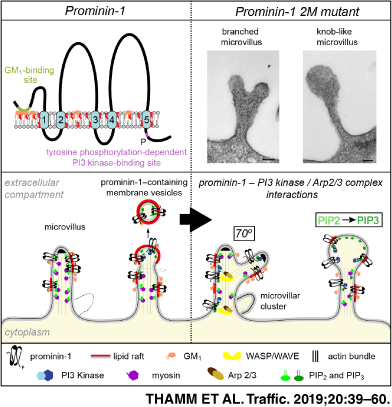
Prominin-1 comprises five transmembrane segments that link an N-terminal extracellular domain, two short intracellular and two large glycosylated extracellular loops and an intracellular C-terminal domain.7, 8 It is subjected to alternative splicing, which creates a substantial amount of splice variants.2, 3, 20, 21 The functional significance of this variability is not yet understood.22
At the molecular level, prominin-1 is associated with cholesterol-dependent membrane micro/nanodomains (often referred to as lipid rafts).23 Therein, it colocalizes with monosialotetrahexosylganglioside (GM1), which is consistent with the presence of two ganglioside-binding sites in the sequence of prominin-1.24-26 The direct binding of prominin-1 to membrane cholesterol determines its specific microvillar retention, and the cholesterol content regulates the shape of microvilli as well as the budding and fission of prominin-1+ vesicles thereof.23, 27 In addition to the “lateral” binding of prominin-1 to membrane lipids, its “vertical” interactions are modulated by the phosphorylation of tyrosine residue 819 (referring to prominin-1 splice variant 1) by Src and Fyn tyrosine kinases.28 Phosphorylated prominin-1 interacts with the 85-kDa regulatory subunit of phosphoinositide 3-kinase (PI3K) leading to the activation of PI3K/Akt pathway.29 The locally activated PI3K can convert phosphatidylinositol(4,5)-bisphosphate (PIP2) into phosphatidylinositol(3,4,5)-trisphosphate (PIP3) in the inner leaflet of the plasma membrane. PIP2 and PIP3 are known to regulate the interaction of actin-binding and signaling proteins,30-32 thereby possibly inducing the reorganization of cellular protrusions.
After two decades of research on prominin-1 its physiological function remains elusive. Retinal phenotypes in individuals carrying mutations in PROM1 gene and data from murine models hint to a scaffolding role of prominin-1 in the organization and dynamics of membrane protrusions emerging at the base of photoreceptor outer segment.5, 22 The latter is a modified primary cilium. The absence of other major symptoms in patients or phenotypes of Prom-1 null mice can be explained, at least in part, by an upregulation of prominin-2 in tissues where both molecules are expressed. This is not the case in the retina.19, 33-37
Prominins may constitute multivalent modules that, together with their interacting partners, could alter the membrane composition and/or the organization of cellular protrusions.12, 38-40 Here, we investigate this issue by creating specific mutations in prominin-1 that obstruct the interaction with its partners and study their impact on microvilli, which we use as a model of actin-driven membrane protrusions.41 Microvilli consist of a bundle of parallel actin filaments that protrudes from the cell surface while its base is interconnected with a dense meshwork of actin and spectrin known as the terminal web. Our data suggest that the synergy of lipid/protein-prominin-1 complexes regulates the architecture and dynamics of cellular protrusions.
2 RESULTS
2.1 Distribution and morphology of microvilli at the apical plasma membrane
Prior to the analysis of the impact of prominin-1 on microvilli, we performed a high-resolution characterization of the apical membrane of polarized Madin-Darby canine kidney (MDCK) epithelial cells by scanning electron microscopy (SEM). At 6 days post-confluence (dpc), the distribution of microvilli varied considerably between neighboring cells ranging from densely packed to sparsely scattered (Figure 1A). Microvilli were present as individual units or formed clusters comprising about 10 microvilli (Figure 1A). The mechanism underlying the formation of microvillar clusters is poorly characterized.42, 43 No microvillus was detected in the proximity of the primary cilium, while those at cell borders were arranged in an array (Figure 1A). Microvillar length differed from cell to cell and was categorized into short (Figure 1B, <200 nm), intermediate (200-400 nm) and long (>400 nm). Few cells showed membrane ruffles or branching microvilli (Figure 1B). The distribution and morphology of microvilli were uniform throughout the surface of a given cell, but differed between adjacent cells (Figure 1B). No clusters of cells with a particular pattern were recognized, indicating that the clonal origin does not explain the morphological variability.

A quantitative analysis based on SEM data revealed numerous cells with membrane ruffles at early time points of polarization (Figure 1C, 1-3 dpc; Supporting Information Table S1). From 6 to 12 dpc, short and intermediate microvilli were most common. We rarely observed cells harboring long or branched microvilli or membrane ruffles (<3%). Microvillar heterogeneity was less pronounced in subconfluent cultures, in which many cells did not show any membrane protrusions while others developed membrane ruffles and/or short microvilli (Figure S1A-D). Varying numbers of filopodia were detected at free cell borders and microvilli arrays started to form at cell-cell contacts (Figure S1E-G).
2.2 Expression of human prominin-1 in epithelial cells
To study the influence of prominin-1 on microvilli we overexpressed two splice variants of human prominin-1 (s1 and s2) in MDCK cells. They differ by the short facultative exon 3 in the extracellular N-terminal domain of splice variant s2 that encodes for nine amino acids (PETVILGLK; Figure 2A, red square). Although the function of these residues is unknown, they might interfere with the interaction between prominin-1 and the ganglioside GM1. As described by Taïeb et al, prominin-1 contains two glycosphingolipid-binding domains.26 One is located in the N-terminal domain and is proposed to bind GM1 (Figure 2A, orange cylinder) while the other was found in the vicinity of the second transmembrane domain and interacts with GD3 (Figure 2A, yellow cylinder). The GM1-binding site contains typical turn-inducing residues (here, Pro37 and Gly50) and an aromatic one (Tyr41). The latter is surrounded by charged amino acids and exposed to the extracellular milieu (Figure 2A and Figure S2A; for details see Reference 26). To determine the impact of this lipid-binding domain on the function of prominin-1 we mutated two of the key residues (Pro37 → Ala and Tyr41 → Ser) in both splice variants (Figure 2A). They were termed 2M mutants. These residues are conserved among other mammalian prominin-1 sequences (Figure S2A). Upon stable transfection, more than 85% of the cells of each cell line expressed the recombinant protein as determined by flow cytometry (Figure S3A) with comparable expression levels as observed by immunoblotting (Figure 2B and Figure S3B,C).

The localization of prominin-1 was then examined by double labeling with mouse monoclonal antibody (mAb) CD133/1 directed against human prominin-1 and fluorescent cholera toxin B subunit conjugates that recognize ganglioside GM1. Confocal laser scanning microscopy (CLSM) analysis revealed the colocalization of both prominin-1 splice variants with GM1 at the apical membrane (Figure 2C, top panels and Figure S2B), in agreement with our previous report.25 The punctate pattern is characteristic for prominin-1 associated with microvilli.23, 44 Both 2M mutants also colocalized with GM1 indicating that the created point mutations did not interfere with the proper microvillar localization of prominin-1 (Figure 2C, bottom panels and Figure S2B).
2.3 Prominin-1 and its mutants increase the number of microvilli
Does prominin-1 influence the architecture of microvilli? To address this question we analyzed the apical plasma membrane using transmission electron microscopy (TEM) and SEM. The analysis suggested that both splice variants of prominin-1, notably their 2M mutants, increase the number of microvilli (Figure 3A and Figure S4A). Besides, we noticed the occurrence of intracellular multivesicular structures in cells expressing 2M mutants (Figure 3A and Figure S4A, green asterisk).

SEM micrographs were used for the quantification of microvilli and revealed an average number of 95 ± 33 microvilli on wild-type cells (MDCK) for a surface area of 12 μm2. This number was significantly increased in cells expressing human prominin-1, that is, 127 ± 49 on splice variant s1 and 127 ± 36 on s2 (Figure 3A, SEM top panels and Figure 3B). Interestingly, mutations in the GM1-binding site further increased their number (see also Figure S4A). The 2M mutants harbored on average 168 ± 56 (s1) and 156 ± 43 (s2) microvilli. On wild-type MDCK cells, about 10 microvillar clusters were observed for a cell surface area of 17.3 μm2 (Figure 3A, SEM bottom panels and Figure 3C) and each of them comprised about 10 ± 3 microvilli (Figure 3D). On cells expressing prominin-1.s1 or s2, a significant increase in the number of clusters and microvilli per cluster was detected (Figure 3C,D). The 2M mutants did not exert further effect on microvillar clusters, but the number of microvilli per cluster was increased in prominin-1.s1 2M (Figure 3C,D).
2.4 Prominin-1 alters the architecture of microvilli
During our EM investigations we recognized alterations in the shape of microvilli (Figure 4A,B and Figure S4A), which prompted us to conduct a quantitative analysis. Using SEM, more than 500 cells per line were randomly picked and assigned to one of the following categories: short, intermediate, long, branched, knob-like/irregularly shaped microvilli or membrane ruffles (Figure 4C and Table S2). About 25% of prominin-1+ cells showed branched microvilli irrespective of the splice variant (Figure 4A-C, green). Each branched segment was about the same diameter as the original microvillus. This phenotype was scarcely observed in control cells, that is, MDCK wild type or those expressing an empty vector (MOCK) (Figure 4C). The expression of 2M mutants led to a 2-fold increase in cells with branched microvilli (Figure 4C). Moreover, ≈20% of the cells harbored irregularly shaped microvilli often with a knob-like structure at their tips (Figure 4A-C, purple). These microvilli frequently showed branching at the same time indicating that both phenotypes are linked (Figure 4B). The knob-like phenotype was not observed in control cells and only in ≈2 and ≈10% of cells expressing prominin-1.s1 or s2, respectively (Figure 4C). In line with the observation made in wild-type cells, the altered morphologies of microvilli were distributed uniformly throughout the surface of a given cell, but differed between adjacent cells (Figure S4B). The coincidence of altered phenotypes and an increase in the number of microvilli suggests that prominin-1, notably its mutants, did not induce the formation of microvilli but might affect their stability, dynamics and/or their resorption. Indeed, all altered microvilli are of intermediate size indicating that prominin-1, and notably its 2M mutants, increases the length of microvilli.

Cells expressing the 2M mutants also harbored microvilli with small protruding vesicle-like structures at their tips suggesting that the budding of membrane vesicles might be affected (Figure 4A and Figure S4A, asterisk). This prompted us to examine the release of prominin-1+ vesicles. Therefore, cells were incubated for 16 hours with medium, which was then subjected to differential centrifugation followed by immunoblotting for prominin-1 and β-actin (Figure 5A). In cells expressing wild-type protein, the amount of prominin-1 recovered in Vesicle fractions corresponded to ≈10% to 12% of total immunoreactivity (Cell + Vesicle). This number dropped to 5% in both 2M mutants suggesting that the alterations of microvilli affected the proper budding and fission of vesicles (Figure 5B, n = 8).

2.5 PI3K inhibition reduces modified microvilli and restores vesicle release
We hypothesize that the overexpression of prominin-1.s1 or s2, and particularly the 2M mutants, may cause an activation of PI3K, which is an interacting partner of prominin-1. As a result PIP2 might be converted into PIP3 in the inner leaflet of the microvillar membrane. Because scaffolding proteins bind to PIP2 and physically couple the plasmalemma to actin filaments, the phospholipid conversion might favor the detachment of the membrane from the underlying cytoskeleton and explain our phenotypes. To address this issue, cells were incubated for 4 hours with PI3K inhibitor LY294002 or dimethyl sulfoxide (DMSO) as vehicle control. Microvillar phenotypes and the release of prominin-1+ vesicles were evaluated. The inhibition of PI3K caused a decrease in the number of cells with altered microvilli and an increase in those harboring short ones (Figure 6A,B). Solely 11% ± 4% of prominin-1.s2 2M-expressing cells displayed knob-like microvilli upon LY294002 treatment while DMSO-treated cells showed 24% ± 6% (Figure 6B, purple). Cells with branched microvilli were also reduced. Similar data were obtained with splice variant 1 (Figure 6B and Table S3).

In 2M mutants-expressing cells treated with LY294002, the reduction of altered microvilli was reflected by a 3-5-fold increase in the release of prominin-1+ vesicles in comparison to DMSO alone (Figure 6C). No significant increase was observed in cells expressing wild-type prominin-1 (Figure 6C). Of note, DMSO caused the formation of short microvilli at the expense of intermediate ones (Figure 6B and Table S3). DMSO did not stimulate the release of prominin-1+ vesicles, suggesting that the occurrence of short microvilli is not the result of extended release (data not shown, see below).
2.6 Arp2/3 complex inhibition reduces modified microvilli
Using SEM micrographs, we evaluated the angle formed by two branching protrusions within a given microvillus. The observed angle of 64.7° ± 12.5° (n = 21) was highly reminiscent of the activity of Arp2/3 complex that choreographs the formation of branched actin networks.45 To evaluate its role, we incubated 2M mutants-expressing cells for 16 hours with the Arp2/3 complex inhibitor CK-666 or its inactive analogue CK-689.46 No differences were observed between cells treated with CK-689 or DMSO as control (Figure 7A,B). However, cells incubated with CK-666 showed a significant reduction of branched and knob-like microvilli, suggesting that the Arp2/3 complex is involved in the alteration of microvilli (Figure 7A,B and Table S4).

Given that the individual inhibition of PI3K and Arp2/3 complex impaired the formation of both branched and knob-like microvilli we evaluated the combined effect. The incubation of cells with LY294002 and CK-666 created a synergistic effect that almost abolished altered microvilli (Figure 7A,C and Table S5). The number of individual microvilli and clusters of them were also significantly reduced reaching a level comparable to wild-type cells under the same conditions, whereas the number of microvilli per cluster remained similar (Figure 7D-F). Thus, the increased number of microvilli in cells expressing prominin-1 mutants can be explained by their morphological alteration, which might interfere with their turnover. The altered microvilli generated by the overexpression of wild-type prominin-1.s1 and s2 were also lost upon the combined inhibition of PI3K and Arp2/3 complex (Table S5). Likewise, we detected a significant reduction in the number of microvillar clusters in non-transfected cells, which also suggests that their formation relies on the activity of PI3K and Arp2/3 complex (Figure 7E). Collectively, these data suggest that the overexpression of wild-type prominin-1 as well as mutations in its GM1-binding site induce a crosstalk with the PI3K pathway and the Arp2/3 complex, and hence a remodeling of the actin cytoskeleton.
2.7 Impaired prominin-1-PI3K interaction reduces altered microvilli and restores vesicle release
To evaluate the impact of direct interaction between prominin-1 and PI3K on microvillar structure, we generated an additional mutation in the 2M mutants at tyrosine 819/828 (with reference to prominin-1.s1 and s2, respectively; Figure 2A and Figure S2A). This amino acid is found in a tyrosine phosphorylation consensus site [R/K]xxx[D/E]xxY,28 which is conserved in all prominin-1 sequences reported so far (Figure S2A). The mutation impedes the phosphorylation of prominin-1 and thereby its interaction with the 85-kDa regulatory subunit (p85) of PI3K. The changes in tyrosine phosphorylation of cell surface prominin-1 were demonstrated by its immunoisolation using AC133 mAb-conjugated magnetic beads and immunoblotting with anti-phosphotyrosine Ab (Figure 8A). The quantification revealed a significant reduction in the phosphorylation of 2M Y819/828F mutants (Figure 8B). The remaining phosphotyrosine signal can be attributed to the second phosphorylation site in prominin-1 at tyrosine 843/852.28 Interestingly, we detected only a slight but not significant increase in the phosphorylation of tyrosines in 2M mutants compared with their wild-type proteins. Nonetheless, the interaction of prominin-1 with p85 was strongly stimulated in cells expressing 2M mutants, but not in those carrying the additional tyrosine mutation (Figure 8C,D). These data suggest that mutations in the first ganglioside-binding site of prominin-1 influence its interaction with PI3K. This observation together with the impact of the Arp2/3 complex inhibitor on microvillar structure prompted us to probe the collected materials for members of the Arp2/3 complex, that is, Arp2 and p34-Arc/ARPC2. Surprisingly, the two subunits were co-immunoisolated with prominin-1 (Figure 8C,D). Again, the 2M mutations increased these interactions while the additional tyrosine mutation reduced them (Figure 8D).

Morphologically, quantitative SEM analysis revealed that the amount of prominin-1 2M Y819/828F-expressing cells with knob-like/irregular microvilli dropped to 2% to 5% (Figure 9A,B). The number of cells harboring branched microvilli was reduced by half in comparison to 2M mutants (Figure 9B). These data indicate that the lack of tyrosine 819/828 phosphorylation impedes the structural alterations provoked by mutations in the GM1-binding site. As a result, cells expressing prominin-1 2M Y819/828F show a distribution of microvillar phenotypes similar to their corresponding wild-type proteins (Figures 4C and 9B). The additional Y819/828F mutation also restored the release of prominin-1+ vesicles (Figure 9C).

2.8 Ubiquitination of prominin-1 and its interaction with syntenin-1 are stimulated in 2M mutants
As microvilli stability and/or dynamics are changed upon the mutations in prominin-1 we suspected that its recycling and degradation might be altered.14, 47 Therefore, we evaluated whether prominin-1 is subjected to ubiquitination and/or interacts with the adaptor protein syntenin-1.16 Both, the ubiquitination of cell surface prominin-1 and its interaction with syntenin-1, were significantly increased in cells expressing 2M mutants suggesting that mutations in the ganglioside-binding site of prominin-1 can trigger its recycling. The additional Y819/828F mutation reduced both interactions (Figure 8E,F). In contrast, no interaction with ezrin, an adaptor protein of the ezrin/radixin/moesin (ERM) family, was observed (Figure 8E), which is in line with our previous study.16
2.9 Mutating cytoplasmic tyrosine 819/828 in prominin-1 results in short microvilli
Does tyrosine phosphorylation of prominin-1 per se affect the microvillar architecture? To address this question, we analyzed their morphology in cells expressing human prominin-1.s1 or s2 carrying solely the Y819/828F mutation. This mutation impeded the phosphorylation of prominin-1 and its interaction with the p85 subunit of PI3K and Arp2/3 complex (Figure 8A-D). CLSM analysis revealed that both mutants localized to the apical plasma membrane, but the immunoreactive signal of prominin-1 appeared narrower in the mutants in comparison to wild-type protein (Figure 10A, right panels). SEM analysis revealed that ≈75% of cells expressing mutant proteins displayed short microvilli at 6 dpc (Figure 10B,C and Table S6) and 14 dpc (Table S6), which is a significant increase compared with control cells (P < 0.005). This suggests that the phosphorylation of prominin-1 and/or its interaction with PI3K affects the length of microvilli. Moreover, no altered microvilli were detected suggesting that the point mutation neutralizes the effect of prominin-1 overexpression. These data are in line with those obtained upon PI3K inhibition. Finally, the Y819/828F mutation did not stimulate the release of prominin-1+ vesicles, indicating that short microvilli are not the result of extensive membrane budding (Figure 10D, see also Figure 5B).

In sum, our data suggest that multiple interactions of phosphorylated prominin-1 with regulatory proteins, such as PI3K and the Arp2/3 complex, are implicated in the architecture and dynamics of microvilli on the apical surface of epithelial cells.
2.10 Prominin-1 mutants alter membrane protrusions of fibroblast-like cells
Given that the expression of prominin-1 is not limited to epithelial cells we transfected CHO cells, which have fibroblast-like characteristics, with prominin-1.s2 and its corresponding mutants. Interestingly, the expression of prominin-1.s2 increased the length of filopodia emerging from the cell border. Precisely prominin-1.s2-expressing cells showed a length of 3.72 ± 1.41 μm (mean ± SD, more than 1000 filopodia were analyzed) vs 1.71 ± 0.70 (n > 300) and 2.24 ± 0.96 (n > 600) μm of untransfected (CHO) and green fluorescence protein (GFP)-transfected cells, respectively (P < 0.01, two-tailed unpaired Student's t-test; Figure 11A, see cartoon). To visualize the cell surface, cell lines were labeled with fluorochrome-conjugated wheat germ agglutinin (WGA).

Moreover, filopodia of prominin-1.s2-transfected cells were often branched, and we observed the appearance of narrow, sheet-like structures morphologically similar to growth cones (Figure 11A,B, red asterisk).48 Often, long filopodia emerged from the apex of these structures. Growth cone-like structures were barely detected in prominin-1-negative cells. Astonishingly, they were more abundant in prominin-1.s2 2M mutant-expressing cells and the length of filopodia was also further increased (5.52 ± 2.27 μm, n > 600). Cells expressing prominin-1.s2 2M Y828F developed numerous axial and radial filopodial branches emanating from the growth cone-like structures. The latter appeared larger than those of non-mutated or prominin-1.s2 2M mutant cells. The length of filopodia of prominin-1.s2 2M Y828-expressing cells was 5.49 ± 2.04 μm (n > 180). In contrast, cells transfected with prominin-1.s2 carrying only the Y828F mutation showed short filopodia emanating from growth cone-like structures and lamellipodia (Figure 11A,B). Filopodia growing out from the cell border were of similar length as those developed by cells expressing wild-type prominin-1, that is, 4.14 ± 2.03 μm (n > 600), but statistically shorter than those of 2M mutant cells (P < 0.05). In all examined cells, the preferential localization of prominin-1 in highly curved membranes (eg, in filopodia and at the edge of the cell border) is evident.
In contrast to the vertical apical microvilli of MDCK cells, the horizontal formation of filopodia on a planar surface facilitated the investigation of the localization of the p34-Arc/ARPC2 subunit of the Arp2/3 complex. In cells expressing GFP (MOCK) or prominin-1 Y828F, the p34-Arc/ARPC2 staining appeared more distant to the edge of the cell membrane and weaker compared with the staining of those expressing prominin-1 wild-type or 2M mutants (Figure 11C, dashed line). In the latter, a colocalization of prominin-1 and p34-Arc/ARPC2 was observed (Figure 11C, white arrowheads) and p34-Arc/ARPC2 was concentrated in the growth cone-like structures (Figure 11C, red asterisk). However, p34-Arc/ARPC2 is excluded from filopodia in all cell lines except the one expressing prominin-1 2M Y828F, which could explain the presence of highly branched filopodia (Figure 11C, red arrowhead). It is interesting to note that the colocalization of prominin-1 and p34-Arc/ARPC2 was also detected in the “soma” part of the cells, particularly in those expressing prominin-1 2M Y828F (Figure 11C, white arrows).
Using our co-immunoisolation protocol (see above), we observed an increased interaction of prominin-1.s2 2M mutant with Arp2/p34-Arc/ARPC2 subunits of the Arp2/3 complex in comparison to wild-type prominin-1 and Y828F mutant (J.K. and D.C., unpublished data). The same observations were made with PI3K (ie, p85; data not shown). Altogether, prominin-1 influences the structure of filopodia and the localization of the p34-Arc/ARPC2 subunit of the Arp2/3 complex in fibroblast-like cells. Furthermore, the data suggest that this interaction, as well as the one with PI3K, occur in both polarized epithelial and fibroblast-like cells.
2.11 Silencing of prominin-1 alters microvilli of HSCs
All data presented above suggest that mutations in human prominin-1 influence the structure of microvilli and other membrane protrusions. This prompted us to investigate the microvillar morphology of a new murine model (rd19) where a spontaneous mutation in Prom1 gene causes prominin-1 knockdown.34 As demonstrated by immunoblotting, both N-glycosylated prominin-1 found in the kidney as well as its endo H-resistant or PNGase F-sensitive deglycosylated forms were not detected in rd19 mice (Figure 12A). TEM analyses of the kidney and duodenum revealed no major alterations of individual microvilli present in both epithelia (Figure 12B,C, respectively), which is in line with previous observations in another prominin-1 null mice.37 The length, diameter and internal actin cytoskeleton of microvilli are not markedly affected by the lack of prominin-1 (Figure 12C). However, we observed budding-like structures at the basal part of microvilli more often in rd19 mice than in wild-type animals, where they were detected at the tip of microvilli (Figure 12C, red and black arrows, respectively). The latter might generate extracellular membrane vesicles while the former can result in both, vesicles and branched microvilli. Interestingly, the organization of the apical cortex of certain rd19 duodenal cells seemed uneven, which led to the appearance of cluster of microvilli (Figure 12D, dashed line) with fused and/or branched microvilli (Figure 12D, asterisks, red dots). Although fused microvilli can be observed in wild-type cells they were more pronounced in cells lacking prominin-1. The co-expression of prominin-2 in the kidney and small intestine, including the duodenum,19, 33-37 might explain the lack of general phenotype (see Section 3).

Given that MDCK cells express both canine prominin-1 and prominin-249 (K.T. and D.C., data not shown), we decided to study the consequences of the lack of human prominin-1 in HSCs. These cells express solely prominin-1.s27, 8 and, importantly, are devoid of prominin-2.19, 33-37 To that end, primary human CD34+ HSCs were immunoisolated from patients and prominin-1 was knocked down using short interfering (si) RNA oligonucleotides as described previously.50, 51 Cells were then cultured on primary mesenchymal stromal cells (MSCs), which were used as feeder cell layer. MSCs secreted various soluble factors, including stromal cell-derived factor-1, and thus mimic to a certain degree the physiological conditions associated with the bone marrow niche.51 We have previously demonstrated that the silencing of prominin-1 did not affect the migration and general polarization of CD34+ HSCs, that is, the formation of the uropod at the rear and lamellipodia at the leading edge.50 At that time, we did not determine the impact on microvillus-like structures, particularly those associated with the uropod where prominin-1 is concentrated.51 ,52 Strikingly, SEM analysis revealed that the number of microvillus-like per uropod was significantly reduced in cells treated with siRNA against prominin-1 in comparison to cells transfected with control-siRNA (Figure 13A,B). In contrast, the knockdown of CD82, a tetraspanin protein associated with the HSC-uropod,52 did not affect microvilli found thereon (Figure 13A,C).

3 DISCUSSION
In this study, we deciphered the impact of prominin-1 on the architecture of microvilli used as a model of actin-driven membrane protrusions.53 Each microvillus contains an array of 20-30 parallel actin filaments that provides structural support. Within a cell, microvilli are typically homogeneous in length and width.54 For our investigations, we overexpressed two distinct splice variants of human prominin-1 and created mutations in their extra- and intracellular domains that would impede their interactions with ganglioside GM1 (2M mutants) and PI3K (Y819/828F mutants), respectively.26, 28, 29
The expression of both splice variants increased the number of individual microvilli and clusters of them. Mutations in their ganglioside-binding site further raised the amount of individual microvilli. The ultrastructural analysis and drug treatments revealed that prominin-1 might not promote the formation of microvilli per se, but could affect their turnover due to alterations in their architecture.55-57 Two major phenotypes were observed: branched microvilli and irregular ones with a knob-like structure at the tip. Both are enhanced in cells expressing the 2M mutants. Concomitant with this, the release of prominin-1+ vesicles was impaired suggesting that the dynamics of microvilli were perturbed.17, 27, 58
Although poorly characterized, branched microvilli have been described in the literature, for example, in rat mammary ascites adenocarcinoma cells,59 human conjunctival epithelium60, 61 and principal cells of rat epididymis.62 They were also observed in diseases such as microscopic enteritis and celiac disease.63, 64 Burgess and Grey reported the induction of branched microvilli by cytochalasin B treatment, which also perturbed their spatial distribution over the apical surface of cells. This suggests that a defect in the organization of actin filaments might also affect the actomyosin network at the base of microvilli65 that provides the force to maintain the organization of the apical cortex.66 At early stages of cell polarization, this network together with actin-remodeling proteins possibly regulates the dynamics of plasma membrane protrusions (eg, membrane ruffles) leading to the formation of individual microvilli and/or clusters of them. This might also explain why a given microvilllar phenotype is detected throughout the entire cell surface. Likewise, the observed increase in short microvilli after DMSO treatment could be caused by negative effects on microfilament organization.67 Surprisingly, DMSO did not perturb microvilli of cells expressing prominin-1.s2 2M, suggesting that its expression modified the membrane-cytoskeleton organization.
The results of the analysis of rd19 knockdown mice are very interesting in this context. These animals are viable and fertile and did not develop major abnormalities other than a progressive photoreceptor degeneration leading to the complete loss of vision as reported for other models of prominin-1-deficient mice.34 Although prominin-1 is strongly expressed in the kidney and duodenum brush border membranes,7, 33-37 the general organization of individual microvilli and epithelia appeared normal. Nonetheless, we could observe budding-like structures at the basal part of microvilli that could result in the release of small membrane vesicles or the formation of branched microvilli. The apical cortex of some cells within duodenal epithelium was also altered leading to the formation of diverse clusters of microvilli with individual microvilli harboring branches. These alterations are in line with an effect of prominin-1 on membrane-cytoskeleton organization of the apical cortex. The coexpression of prominin-2 in these tissues19, 33-37 might explain the lack of a general phenotype as demonstrated in the adult murine hippocampus where an upregulation of prominin-2 compensates for prominin-1 deficiency.36 Further investigations, for example, the use of serial section EM and/or double knockout mice, will help to decipher the impact of prominin-1 and -2 on the terminal web throughout the polarization process of epithelial cells. In non-epithelial cells expressing solely prominin-1 such as CD34+ HSCs, its silencing demonstrated its implication in the formation and/or maintenance of microvillus-like protrusions. The latter protrusions might be more dynamic then those in the brush border membrane and explain the direct impact of prominin-1. The physiological significance of the lack of these finger-like protrusions at the uropod of the blood stem and progenitor cells remains to be explored.50, 51
Pharmacological interference, co-immunoisolation and specific mutations in prominin-1 revealed that the nucleation of branched microvilli is driven by the Arp2/3 complex and its interaction with prominin-1. Arp2/3 complex induces the formation of branched actin networks at the leading edge of migrating cells, in neuronal growth cones and in the precursor structures of dendritic and axonal filopodia (see below). 68-70 To our knowledge, it is not yet described to play a role in microvilli. Indeed, the Arp2/3 complex as well as its activator Wiskott-Aldrich syndrome protein (WASP) seem to be dispensable for the formation of mammalian microvilli,71-73 but they play a role in sheet-like protrusions and the initiation of rhabdomere microvilli in the fruit fly.74, 75 They are also required for the apical trafficking of certain membrane proteins into microvilli.76 The significant reduction of microvillar clusters on drug-treated cells strongly suggests that the Arp2/3 complex, together with prominin-1, is involved in their biogenesis (see below). Thus, the interaction of prominin-1 with actin-remodeling proteins might be relevant for (a) the formation of membrane ruffles at early stages of polarization; (b) the biogenesis of microvillar clusters and (c) the recycling of prominin-1 in the endocytic pathway. The increase in ubiquitination of prominin-1 and its enhanced interaction with syntenin-1 in cells expressing 2M mutants suggest a high endocytic turnover.
Beyond microvillar structure, induction of actin filament branching by prominin-1 is consistent with its localization at the leading ledge of lamellipodia and magnupodia of migrating progenitor cells or cancer cells.14, 77 These protrusions are the driving force for cell migration under normal and pathological conditions and they are powered by actin polymerization. The latter is mediated by Arp2/3-induced nucleation of branched actin networks and the elongation of actin filaments. Thus, prominin-1-induced actin filament branching might explain the high metastatic capacity of prominin-1+ cancer cells and their reduced migration upon prominin-1 silencing.78, 79 Moreover, it is well established that controlled and directional cell movement requires local production of PIP3 by PI3K that indirectly actives WASP-family and verprolin-homologous (WAVE) family proteins, which in turn triggers the activation of Arp2/3 complex leading to lamellipodia formation.80-82 In this context, the phosphorylation of prominin-1 and its interaction with the PI3K can play a leading role,29 and therefore our findings could make a significant impact on the treatment of metastatic cancer cells with the prominin-1-Arp2/3 complex axis as key target in novel anti-invasive therapies.83 All observations (and hypotheses) were recapitulated by the data obtained from studying CHO cells where numerous structural alterations of the plasma membrane were observed upon the expression of prominin-1 and its mutants. The induction of sheet-like structures that are reminiscent of neuronal growth cones located at the tips of neurites as well as branched filopodia are two examples. Indirectly, our data suggest that prominin-1 itself, its interaction with GM1/PI3K/Arp2/3 complex, and/or its active recycling (see above) may exert an impact on the formation of neuronal growth cones. These thrilling issues request further investigations. Of note, the colocalization of prominin-1 and p34-Arc/ARPC2 also requires further attention, particularly in regard to the implication of prominin-1 in the formation and/or maintenance of focal adhesions. These multiprotein structures form mechanical links between intracellular actin bundles and the extracellular matrix. Their regulation by certain subunits of Arp2/3 complex, notably p34-Arc/ARPC2 and vinculin, has been proposed.84 Similarly, Liu et al have demonstrated that the interaction of prominin-1 with Src protein promotes the phosphorylation of focal adhesion kinase and cancer cell migration.85
Do the subunits of Arp2/3 complex and PI3K directly interact with prominin-1 as suggested by co-immunoisolation? It is known that the scaffold protein IQ motif containing GTPase activating protein 1 (IQGAP1) together with Arp2/3 complex promotes the nucleation of branched actin networks and provides the force for lamellipodial protrusions and cell migration.86, 87 Thus, IQGAP1 may mediate the formation of a multiprotein complex including all aforementioned proteins, actin and WAVE proteins.86 Excitingly, the proper positioning of these players along actin microfilaments can be regulated by PIP3.88 Supporting this hypothesis, we found that prominin-1 and IQGAP1 are coimmunoisolated from MDCK and CHO cells in a pattern similar to p85/Arp2/p34-Arc/ARPC2 with respect to wild-type and mutant prominin-1 (J.K. and D.C., unpublished data). Altogether, the multiple interactions of prominin-1 imply its association with various cellular processes as reported for IQGAP1 (reviewed in References 86, 89).
The observed knob-like phenotype of microvilli has been previously reported for endothelial cells of cerebral arteries from spontaneously hypertensive rats and upon the action of insulin on rat hepatocytes.90, 91 The latter observation was related to PI3K activity.92 The interaction of prominin-1 with the PI3K can explain the formation of knob-like microvilli, its link to the branching phenotype and the dynamics of vesicle release. Precisely, the microvillar membrane is coupled to actin filaments by linker proteins such as myosin and ezrin, and their interactions are regulated by clusters of PIP2 (Figure 10E, model s1/s2).93-96 The conversion of PIP2 into PIP3 in the inner leaflet of the plasma membrane by a prominin-1-driven PI3K activation would favor the decoupling of the membrane from the underlying cytoskeleton (Figure 10E, model 2M) resulting in microvilli with irregular shapes. This would also impede the release of prominin-1+ vesicles. The increase in PIP3 might also induce the activation of the Arp2/3 complex thereby linking knob-like microvilli to the branched phenotype.
How can mutations in the extracellular domain of prominin-1 influence the activity of cytosolic proteins, and hence microvillar organization? And why does the structural impact of prominin-1.s1 (and its 2M mutant) seem to be less severe than the one of s2 variant? The amino acids encoded by exon 3, which is found only in splice variant s2, might act as a spacer element that regulates the interaction of prominin-1 with gangliosides. This interaction within the outer leaflet may determine the phospholipid composition in the inner leaflet (eg, PIP2/PIP3 ratio) by regulating the prominin-1-driven PI3K activation (Figure 10E). Therefore, by promoting membrane lipid clustering prominin-1 may mediate a direct crosstalk between the outer and inner leaflet of the plasma membrane. Its lipid interactors may also control the shape of highly curved membranes such as those found in microvilli, cilia, filopodia, membrane vesicles and the leading edge of lamellipodia.38, 39 Interestingly, cholesterol depletion also influences the release of prominin-1+ vesicles and produces microvilli with pearling shapes similar to those observed in cells expressing prominin-1 2M mutants.27 Besides, both membrane cholesterol-depleted cells and those expressing prominin-1 2M mutants harbor numerous intracellular multivesicular structures suggesting a defect in intracellular trafficking.27
In a similar context, Ikenouchi et al have elegantly demonstrated that the proper microvillar structure requires sphingomyelin clusters that induce co-clustering of the membrane protein podocalyxin-1, which recruits the adaptor protein ERM-binding phosphoprotein of 50 kDa (EBP50), ERM proteins and phosphatidylinositol 4-phosphate 5-kinase β leading to the local accumulation of PIP2.97 Our results show that a direct interaction of prominin-1 with ERM protein ezrin can be excluded, but the distribution of ERM proteins in prominin-1-expressing cells has to be investigated.
Unexpectedly, the phosphorylation of prominin-1 that mediates its interaction with PI3K seems to be necessary for the elongation of microvilli pointing to the importance of a dynamic equilibrium between PIP2 and PIP3 (Figure 10E, model Y819/828F). It remains to be determined whether the lack of phosphorylation of the tyrosine residue 819/828 of prominin-1 directly interferes with actin-bundling or -capping proteins, such as epidermal growth factor receptor pathway substrate 8 (Eps8).43
Despite these findings several issues remain to be explored. The significance of the second ganglioside-binding site (ie, GD3) and the existence of multiple prominin-1 splice variants (up to 23 until now) will need to be dissected with regard to their specific cellular expression (eg, oligodendrocyte, photoreceptor, stem cell) and their subcellular localization in various types of protrusion (eg, myelin, connecting cilia, motile cilia). In the latter context, we noticed alterations in primary cilia of MDCK cells expressing prominin-1.s1/s2 mutants, notably Y819/828F (K.T. and D.C., unpublished data) and the collapse of motile cilia in ependymal cells where cilium-associated prominin-1 is delocalized in a mouse model of type 2 diabetes.98
Collectively, our investigation suggests that the synergy of prominin-1-lipid/protein complexes regulates the architecture and dynamics of microvilli. This strengthens the role of prominin-1 as an organizer of cellular protrusions. The widespread expression of prominin-1 and its multiple lipid and protein interactions might explain its association with numerous cellular processes that involve a remodeling of the plasma membrane notably proliferation, differentiation, migration, autophagy and carcinogenesis.
4 MATERIALS AND METHODS
4.1 Animals and ethics statement
The prominin-1 knockout mouse (Prom1 rd19) carrying a spontaneous point mutation in Prom1 gene99 was obtained from The Jackson Laboratory (Sacramento, CA). The C57BL/6J mouse strain was used as control. Animal experiments were performed in strict accordance with German Animal Welfare legislation, and all procedures were performed in compliance with the UK Animals (Scientific Procedures) Act 1986, which were approved by the Institutional Animal Welfare Officer. All efforts were made to minimize the suffering of animals, and the ones for organ removal were sacrificed according to the applicable European and National regulations (Tierschutzgesetz). The Max Planck Institute of Molecular Cell Biology and Genetics holds necessary licenses (74-9165.40-9-2000-1 and 74-9168.25-9-2001-1) for keeping and breeding laboratory animals and collecting organs and tissues, both issued by the Regierungspräsidium Dresden, Saxony.
4.2 Plasmid construction
Expression plasmids (hprom-1.s1 and hprom-1.s2) were constructed by introducing the full-length coding sequence of human prominin-1 splice variant 1 (GenBank AF507034.1, cDNA clone MGC: 20041 IMAGE: 4644690) and 2 (AF027208.1; Reference 100) into the enhanced yellow fluorescent protein (EYFP)-N1 vector (BD Bioscience Clontech). XhoI and NotI restriction sites were used for directional cloning.100 Note that XhoI/NotI digestion of pEYFP-N1 vector causes the excision of the fluorescent protein.
4.3 Site-directed mutagenesis
Two amino acids (proline 37 and tyrosine 41) in the GM1-binding site of prominin-126 and tyrosine 819/828 (referring to prominin-1.s1 and s2, respectively) in its cytoplasmic C-terminal end were mutated using QuikChange II Site-Directed Mutagenesis Kit (catalog number #200523; Agilent Technologies) according to the manufacturer's instructions. The hprom-1.s1 and hprom-1.s2 plasmids were used as templates. The applied forward and reverse oligonucleotides were 5′-GGAATTATGAATTGGCTGCAACAAATTCTGAGACCCAAGACTCC-3′ [kt-5′2M] and 5′-GGAGTCTTGGGTCTCAGAATTTGTTGCAGCCAATTCATAATTCC-3′ [kt-3′2M] and 5′-GGATTCGGAGGACGTGTTCGATGATGTTG-3′ [kt-5′Y828F] and 5′-CAACATCATCGAACACGTCCTCCGAATCC-3′[kt-3′Y828F], respectively. In the 2M mutant, two single nucleotide changes (underlined) led to the encoding of alanine (Ala37) and serine (Ser41) instead of proline and tyrosine, respectively. In the Y819/828F mutants, the single nucleotide change (underlined) resulted in the encoding of phenylalanine instead of tyrosine. Expression plasmids were sequenced to confirm the absence of any reading mistakes introduced by the DNA polymerase and/or subcloning steps. Sequencing reactions were performed using Applied Biosystems 3730 Genetic Analyzer at the Sequencing Facility of the Max Planck Institute of Molecular Cell Biology and Genetics (MPI-CBG, Dresden, Germany).
4.4 Cell culture
MDCK cells (strain II, American Type Culture Collection, #ATCC CCL-34) were cultured in Dulbecco's modified Eagle medium (DMEM, 1×, 1 g/L d-glucose, l-glutamine, pyruvate, GIBCO, Thermo Fisher Scientific) supplemented with 10% fetal calf serum (FCS; PAA, GE Healthcare), 100 U/mL penicillin and 100 μg/mL streptomycin (GIBCO), 10 mM HEPES and MEM non-essential amino acids (1×, PAA). CHO cells7 were cultured in Ham's F-12 nutrient mixture (+l-glutamine, GIBCO) containing 10% FCS and 100 U/mL penicillin and 100 μg/mL streptomycin. Both cell lines were maintained in a humidified incubator at 37°C under 5% CO2 atmosphere as described previously.49
Primary human HSCs and MSCs were collected from healthy donors after informed consent and approval of the local ethics committee (ethic board nos. EK201092004 and EK263122004). Isolation and cultivation of MSCs were performed as described.77 CD34+ HSCs were isolated from mobilized peripheral blood directly after leukapheresis by immunomagnetic separation using the magnetic-activated cell sorting system with CD34 MicroBeads (CD34 MicroBead Kit, #130-046-703; Miltenyi Biotec, Bergisch Gladbach) and LS columns following the manufacturer's instructions, as reported previously.77 Mobilization was achieved by subcutaneous injection of 7.5 μg/kg granulocyte colony-stimulating factor per day for 5 days (Granocyte; Chugai Pharma). Freshly isolated CD34+ HSCs were transfected (see below) and cultured on MSCs in HSC medium (serum-free medium [Cell Gro SCGM; CellGenix]) supplemented with early-acting cytokines (50 ng/mL stem cell factor, 50 ng/mL fms-related tyrosine kinase-3 ligand [CellGenix] and 15 ng/mL interleukin-3 [R&D Systems]) at a density of 15 x 104/cm2 for 2 days in a humidified 5% CO2 atmosphere at 37°C.
4.5 Plasmid and siRNA transfections
MDCK and CHO cells were stably transfected with expression plasmids described above by electroporation using Amaxa Nucleofector Kit L (MDCK cells) or T (CHO cells) according to the manufacturer's instructions (#VCA-1005, VCA-1002; Lonza). As control, cells were either transfected with the expression plasmid only or with one that contained a GFP reporter gene (both referred to as MOCK).
Cells expressing the neomycin resistance gene were selected by introducing 500 μg/mL of Geneticin (G418 Sulfate; Thermo Fisher Scientific) into the culture medium. After selection, cells were further expanded in a medium containing 250 μg/mL of the selective agent. Note that the microvillar morphology did not change when cells were cultured in medium with 0.5 or 1 mM geneticin or without (data not shown). These data rule out a direct effect of the drug on polyphosphoinositides.101, 102
To enrich MDCK and CHO cell populations expressing human prominin-1, cells were sorted using paramagnetic labeling with CD133 MicroBeads (CD133 MicroBead Kit, #130-097-049; Miltenyi Biotec) and LS columns. To enhance the expression of the transgene 10 mM sodium butyrate was added to the culture medium for 16 hours except for SEM and TEM analysis.44 If not stated otherwise, cells were used at 6 dpc. To investigate the influence of the PI3K, MDCK cells were incubated with 5 μM LY294002 (Sigma-Aldrich) or DMSO (1:2000) as negative control for 4 hours prior to their analysis. To investigate the influence of the Arp2/3 complex, cells were incubated with 200 μM CK-666 or its inactive analogue CK-689 (Merck) for 16 hours. Again, DMSO (1:500) was used as control. For combined inhibition of PI3K and Arp2/3 complex, cells were first incubated with CK-666 or CK-689 (200 μM) for 16 hours and, thereafter, with CK-666/CK-689 and LY294002 (5 μM) for additional 4 hours.
Freshly isolated CD34+ HSCs were transfected with siRNA (1 μg) by electroporation using the human CD34 Cell Nucleofector Kit (VPA-1003; Amaxa). Prominin-1 (CD133, ID.HSS113055), CD82 (ID.HSS105654) and negative control (lo GC Duplex; ID.12935-200) Stealth siRNA oligonucleotides were purchased from Invitrogen. After the transfection, cells were washed in pre-warmed medium, spun down, and the cell pellets were incubated for 15 minutes at 37°C in a 5% CO2 humidified atmosphere.51 Transfected HSCs were then distributed onto the MSC monolayer (see above). Under these conditions, the knockdown of targeted proteins was more than 75% as observed by immunoblotting and/or immunofluorescence50 (data not shown).
4.6 Flow cytometry
MDCK cells were harvested with 0.05% trypsin/0.5 mM EDTA solution (Life Technologies) for 15 minutes at 37°C, centrifuged, and the pellet was resuspended in PBS containing 0.2% gelatin and 5 mM EDTA (VWR International). Cell suspensions (2 x 105 cells in 100 μL) were incubated with or without phycoerythrin (PE)-conjugated mAb against human prominin-1 (1:10, clone AC133; Miltenyi Biotec) or human CD34 (1:10, clone 581; BD Bioscience) for 30 minutes at 4°C. Cells were washed with PBS containing 5 mM EDTA. After centrifugation, cells were resuspended and incubated for 10 minutes in 4% paraformaldehyde (PFA). After washing with PBS containing 5 mM EDTA, 20 000 events were acquired on a FACSCanto II System (BD Bioscience). Instrument settings and gating strategies were established using unstained or CD34-labeled cells.
4.7 Differential centrifugation of prominin-1-containing membrane vesicles
Cells were grown in six-well dishes for 6 dpc. After washing with PBS they were supplied with fresh culture medium (2 mL) and incubated for 4 or 16 hours. Afterward, conditioned media were collected, supplemented with Complete protease inhibitor cocktail (Roche Molecular Biochemicals) and subjected to differential centrifugation as follows: 5 minutes at 300g (pellet 1 [P1]); 20 minutes at 1200g (P2); 30 minutes at 10 000g (P3) and 1 hour at 200 000g (P4). Pellets were resuspended in Laemmli buffer. In parallel, adherent cells were harvested and centrifuged for 5 minutes at 300g. The resulting pellets were lysed for 30 minutes at 4°C in solubilization buffer A (1% NP-40, 0.5% deoxycholate, 0.1% sodium dodecylsulfate, 150 mM NaCl, 50 mM Tris-HCl, pH 8.0) supplemented with complete protease inhibitor. Detergent extracts were centrifuged at 4°C (10 minutes, 10 000g) and resulting supernatants were mixed with Laemmli buffer. All samples were analyzed by immunoblotting for prominin-1 and β-actin (see below). For quantification, we combined prominin-1 immunoreactivity associated with cell extract, P1 and P2 fractions and designated them as Cell fraction while materials found in P3 and P4 were referred to as Vesicle fraction.
4.8 Immunoisolation of prominin-1
Cells were grown in T75-flasks for 6 dpc and then solubilized for 30 minutes on ice in solubilization buffer B (0.2% Triton X-100, 150 mM NaCl, 20 mM Tris/HCl, pH 7.5) containing EDTA-free Complete protease inhibitor cocktail (Roche Diagnostics GmbH), 2 mM sodium orthovanadate, 10 mM sodium fluoride (both from New England Biolabs Inc.) and 10 mM β-glycerophosphate (Sigma-Aldrich). After centrifugation (10 minutes, 16 000g, 4°C) for removal of insoluble materials, detergent lysates were divided into two fractions that were incubated for 2 hours at 4°C with 140 μL of either CD133 mAb-conjugated to magnetic beads or anti-fluorescein isothiocyanate (FITC) antibody-coupled magnetic beads as negative control, prior to magnetic separation using MS columns (Miltenyi Biotec). Flow-through materials were collected. Materials retained in columns were washed six times with 1 mL of ice-cold PBS containing 2 mM EDTA and 0.1% Triton X-100 and then flushed with the same buffer. Flow-through and bound fractions were subjected to methanol/chloroform precipitation. Proteins were resuspended in Laemmli buffer and analyzed by immunoblotting for various antigens (see below).
4.9 Endoglycosidase digestion
Kidney lysates were prepared from adult wild-type or rd19 mice using solubilization buffer C (1% Triton X-100, 150 mM NaCl, 20 mM Tris/HCl, pH 7.5) containing Complete protease inhibitor cocktail (Roche Diagnostics GmbH). After 30 minutes on ice, samples were centrifuged (10 minutes, 16 000g, 4°C) to remove insoluble materials. Solubilized proteins (50 μg) were then incubated for 6 hours at 37°C in the absence or presence of either 10 mU endo H (endo-β-N-acetylglucosaminidase H) or 1 U PNGase F (peptide-N-glycosidase F) according to the manufacturer's instructions (Roche Diagnostics GmbH).
4.10 Immunoblotting
Proteins were separated by SDS-polyacrylamide gel (7.5% or 8%) electrophoresis and transferred to polyvinylidene fluoride membranes (pore size 0.45 μm; Millipore Corp.) using a semi-dry transfer cell system (Cti). After transfer, membranes were incubated overnight at 4°C in blocking buffer I (PBS containing 0.3% Tween 20 and 5% milk powder) prior to 1 hour incubation at room temperature (RT) with primary antibody against prominin-1 (1:2000; mouse mAb anti-CD133, clone 80B2584) diluted in blocking buffer I. As loading controls, mouse mAbs AC-74 against β-actin (1:10 000; Sigma-Aldrich) or 6-11B-1 against α-tubulin (1:1000; Sigma-Aldrich) were used. For the analysis of immunoisolated prominin-1, membranes were incubated in blocking buffer II (TBS containing 0.1% Tween 20 and 3% bovine serum albumin) either overnight at 4°C or 1 hour at RT prior to 1 hour incubation at RT with the primary antibodies against prominin-1 (see above), ubiquitin (1:000; rabbit polyclonal [p] Ab; Dako), phosphotyrosine (1:330; mouse mAb, clone PY99; Santa Cruz Biotechnology), p85 subunit of PI3K (1:1000; rabbit pAb; Millipore Corp.), Arp2 (1:250; rabbit pAb [H-84]; Santa Cruz Biotechnology), p34-Arc/ARPC2 subunit of Arp2/3 complex (1:1000; rabbit pAb; Millipore Corp.), syntenin-1 (1:5000; rabbit pAb; Abcam) and ezrin (1:5000; rabbit mAb, clone EP886Y; Abcam). Antibodies were diluted in blocking buffer II. In all cases, antigen-antibody complexes were detected using appropriate horseradish peroxidase-conjugated secondary antibodies (1:5000; Jackson ImmunoResearch Inc.) and visualized with enhanced chemiluminescence reagents (ECL system; GE Healthcare Life Sciences). Membranes were exposed to Amersham Hyperfilms ECL (GE Healthcare). Fiji was used for the quantification of immunoblotted materials.103 Note that none of the current available antibodies directed against human or mouse prominin-1 cross-reacted with canine and/or hamster orthologs.1, 7, 49
4.11 Immunofluorescence and confocal laser scanning microscopy
Cells were grown on fibronectin (BD Biosciences)-coated glass coverslips or in eight-well chambers (μ-Slide 8 Well, #80826; Ibidi) for 6 to 7 dpc. They were incubated with 10 mM sodium butyrate 16 hours before their analysis. For the cell surface labeling, cells were rinsed with Ca/Mg buffer (phosphate-buffered saline containing 1 mM CaCl2 and 0.5 mM MgCl2) and incubated in blocking buffer III (Ca/Mg buffer containing 0.2% gelatin) for 30 minutes at 4°C. Cells were incubated with anti-human prominin-1 mouse mAb CD133/1 (1:50; clone AC133, #130-090-422; Miltenyi Biotec) alone or in combination with Alexa Fluor 488-conjugated cholera toxin subunit B (1:1000; Invitrogen) diluted in blocking buffer III for 1 hour at 4°C. Alternatively, cells were incubated after prominin-1 immunolabeling with FITC- or rhodamine-conjugated WGA (10 μg/mL; Vector Laboratories) for 30 minutes at 4°C. Cells were washed with PBS and fixed in 4% PFA in PBS for 30 minutes at RT. They were then rinsed and quenched for 10 minutes in PBS containing 50 mM NH4Cl prior to the incubation with the appropriate Alexa Fluor 488/546/555-conjugated goat anti-mouse IgG1 (1:500; Molecular Probes, Life Technologies). For the intracellular labeling, PFA-fixed cells were permeabilized with 0.1% saponin diluted in PBS containing 0.2% gelatin (blocking buffer IV)104 for 20 minutes at RT prior to immunolabeling for prominin-1 (see above) and p34-Arc/ARPC2 subunit of Arp2/3 complex using rabbit antiserum (5 μg/mL; Merck). Primary antibodies were observed using Alexa Fluor 488-conjugated goat anti-mouse IgG1 and Alexa Fluor 555-conjugated donkey anti-rabbit IgG. All antibodies were diluted in blocking buffer IV. In some experiments, nuclei were counterstained with 4′-6-diamidino-2-phenylindole (DAPI, 1 μg/mL; Sigma-Aldrich). All samples were then washed three times in PBS containing 0.2% gelatin, two times in PBS and H2O and mounted in Mowiol 4.88 (Merck). Cells seeded in eight-well chambers were not mounted, but directly observed at RT with a confocal Zeiss LSM 700 using ZEN software and a 100× 1.4 oil DIC Plan-Apochromat objective, 4× zoom (Figure 2C and Figure S2B) or a 63× 1.4 oil DIC Plan-Apochromat objective (Figure 10A). All figures were processed with Fiji.103
For the length measurement of filopodia emerging from CHO cells, CLSM micrographs of prominin-1 and/or WGA staining were used. Only cells with spread morphology, and not those of a rounded shape, were taken into consideration. Note that we did not measure the absolute number of their length since they were often tilted. Instead we measured their length from the tip to the planar cell surface with an angle of 90° (see cartoon in Figure 11A).
4.12 Transmission electron microscopy
Culture cells growing on 30-mm dishes were pre-fixed by the addition of 0.1% glutaraldehyde into the cell culture medium and subsequently fixed in 1% PFA, 1% glutaraldehyde in 0.1 M PIPES, pH 7.4 at RT. After washing steps, cells were post-fixed in 1% aqueous osmium tetroxide for 1 hour at RT. After washing with water, cells were incubated with 0.1% tannic acid in water for 10 minutes and contrasted with 1% aqueous uranyl acetate for 30 minutes at RT. The samples were processed through a graded series of ethanol for standard plastic embedding in LX 112 resin (LADD Research Industries). Sheets of resin embedded cells were removed from the dishes by liquid nitrogen. Transverse sections were cut at 70 nm on an UCT ultramicrotome (Leica) and post-stained with uranyl acetate and lead citrate. The samples were viewed in a Morgagni electron microscope (Thermo Fisher Scientific) and images acquired on a Morada CCD camera (EMSIS GmbH).
Adult mice, 4 to 9 months of age, were fixed by perfusion with 2% PFA, 2% glutaraldehyde in 0.12 M phosphate buffer at RT. Tissues were dissected and fixed in the same buffer overnight. After washing steps, the tissue was further dissected into small pieces, which were post-fixed in 1% aqueous osmium tetroxide, 1.5% potassium ferrocyanide for 1 hour at RT. Subsequent steps were performed as described above. Note that Epon replacement (Carl Roth) was used as of resin and embedding was performed in silicon molds.
4.13 Scanning electron microscopy
Samples for SEM analysis were prepared as described.19 Briefly, MDCK cells were grown on fibronectin-coated coverslips for the indicated periods and then fixed in 2% glutaraldehyde overnight at 4°C. The HSC-MSC co-cultures were fixed in the same way. After dehydration in an acetone gradient (25%-100%) all samples were critical point dried in a CO2 system (Critical Point Dryer CPD 300; Leica). Samples were mounted on aluminum stubs, coated with gold (≈20 nm thickness) in a sputter coater (SCD 050; BAL-TEC GmbH) and examined with a SEM (JSM-7500F; JEOL). SEM micrographs were used for the quantification of microvilli of MDCK cells and HSCs.
4.14 Statistical analysis
Statistical analyses were performed using Graph Pad Prism version 6.00 for Mac (Graph Pad Software; www.graphpad.com). Two-tailed unpaired Student's t-test was used for the analysis of the immunoreactivity ratio of prominin-1/β-actin or other antigens, the number of microvilli or microvillar clusters and the prominin-1 immunoreactivity associated with membrane vesicles. The length of filopodia was also compared with this test. Mann-Whitney test was used for the analysis of the number of microvilli per cluster. Two-tailed Fisher's exact test was applied for the evaluation of microvillar phenotypes. The number of events is indicated in the figures and their legends. Observed differences were regarded as significant if the calculated P-values were ≤0.05. *P < 0.05; **P < 0.01; ***P < 0.005.
ACKNOWLEDGMENTS
D.C. was supported by Deutsche Forschungsgemeinschaft (DFG, TRR83, SFB655-B3) and Sächsisches Staatsministerium für Wissenschaft und Kunst (#4-7531.60/29/31). M.B. was supported by DFG (SFB655-B2) and W.B.H. was supported by DFG (TRR83, SFB655-A2).
CONFLICTS OF INTEREST
The authors have no conflicts of interest to report.
Author contributions
K.T., D.S., J.K., W.B.H., M.W.-B. and D.C. conceptualized and designed the project. K.T., D.S., J.K., V.B., D.R., A.M. and M.W.-B. conducted the experiments and analyzed the data. D.C. and W.B.H. analyzed the data. K.T. and D.C. prepared the manuscript. K.T., J.K., M.B., W.B.H., M.W.-B. and D.C. finalized the manuscript. All authors read and approved the final manuscript.
Editorial Process File
The Editorial Process File is available in the online version of this article.

