

A single proline substitution is critical for the thermostabilization of Clostridium beijerinckii alcohol dehydrogenase
Abstract
Analysis of the three-dimensional structures of three closely related mesophilic, thermophilic, and hyperthermophilic alcohol dehydrogenases (ADHs) from the respective microorganisms Clostridium beijerinckii (CbADH), Entamoeba histolytica (EhADH1), and Thermoanaerobacter brockii (TbADH) suggested that a unique, strategically located proline residue (Pro100) might be crucial for maintaining the thermal stability of EhADH1. To determine whether proline substitution at this position in TbADH and CbADH would affect thermal stability, we used site-directed mutagenesis to replace the complementary residues in both enzymes with proline. The results showed that replacing Gln100 with proline significantly enhanced the thermal stability of the mesophilic ADH: ΔT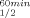 = + 8°C (temperature of 50% inactivation after incubation for 60 min), ΔT
= + 8°C (temperature of 50% inactivation after incubation for 60 min), ΔT = +11.5°C (temperature at which 50% of the original CD signal at 218 nm is lost upon heating between 30° and 98°C). A His100 → Pro substitution in the thermophilic TbADH had no effect on its thermostability. An analysis of the three-dimensional structure of the crystallized thermostable mutant Q100P-CbADH suggested that the proline residue at position 100 stabilized the enzyme by reinforcing hydrophobic interactions and by reducing the flexibility of a loop at this strategic region. Proteins 2007. © 2006 Wiley-Liss, Inc.
= +11.5°C (temperature at which 50% of the original CD signal at 218 nm is lost upon heating between 30° and 98°C). A His100 → Pro substitution in the thermophilic TbADH had no effect on its thermostability. An analysis of the three-dimensional structure of the crystallized thermostable mutant Q100P-CbADH suggested that the proline residue at position 100 stabilized the enzyme by reinforcing hydrophobic interactions and by reducing the flexibility of a loop at this strategic region. Proteins 2007. © 2006 Wiley-Liss, Inc.
INTRODUCTION
One approach to understanding the principles governing the thermostability of proteins is comparing the three-dimensional structures of highly homologous thermozymes and mesophilic enzymes.1-4 Site-directed mutagenesis has been widely used to study many protein functions, including thermostability.5-12 Because universal rules governing thermostability have not yet emerged, families of enzymes having the same biological function should be studied individually.13
Matthews studied the thermal stability of lysozyme using site directed mutagenesis and X-ray crystallography approaches, and proposed that the thermostability of a protein can be increased by selected site-directed substitution of an amino acid that decreases the configurational entropy of unfolding.14
This hypothesis and reports of others12, 15-20 highlighting the unique role of proline in stabilizing local three-dimensional protein structure provided the rationale for studying the effect of proline on the thermostability of alcohol dehydrogenase. We have been studying the molecular basis of thermostability in a model system comprising three functionally related, highly homologous, medium-chain, NADP(H)-linked alcohol dehydrogenases (ADHs; EC 1.1.1.2.), all showing similar substrate specificity.21-27 The enzymes under study are from (a) the extremely thermophilic bacterium Thermoanaerobacter brockii (TbADH),26 first isolated from Yellowstone Park hot springs,24 (b) the mesophilic bacterium Clostridium beijerinckii (CbADH),22 and (c) the protozoan parasite Entamoeba histolytica (EhADH1),23, 27 all of which reversibly catalyze the oxidation of alcohols to the corresponding ketones using NADP(+) as cofactor. We isolated and characterized TbADH and cloned, sequenced, and overexpressed the structural adh genes from T. brockii,28C. beijerinckii,28 and E. histolytica29 in Escherichia coli. Amino acid sequence alignments of the three ADHs (see Fig. 1) revealed high-sequence conservation among the three members of this enzyme family—75% identity between TbADH and CbADH, 66% identity between EhADH1 and TbADH, and 62% identity between EhADH1 and CbADH.5 Interestingly, the proline residues are even more highly conserved than other amino acids in the sequence, with all 13 proline residues of the mesophilic CbADH being present in TbADH, of which 12 are also present in EhADH1.5 Pro 84 and Pro141 in EhADH1 are shifted by one residue from the position of the conserved Pro 83 and 140 of TbADH and CbADH. In addition, the sequence of the extremely thermophilic TbADH contains 8 prolines that are absent in CbADH, whereas EhADH1 contains a unique proline at position 100 that is absent in both TbADH (His100) and CbADH (Gln100). Despite their high degree of sequence homology, the enzymes differ greatly in thermostability, with T1/2 values of CbADH < EhADH < TbADH (64.5°C, 77.5°C, 93.8°C, respectively).5, 28, 30

Amino acid sequence alignment of alcohol dehydrogenases from Thermoanaerobacter brockii (TbADH, TB),26Clostridium beijerinckii (CbADH, CB)28 and Entamoeba histolytica (EhADH, EH).27 Amino acid residues of TbADH are in the one-letter code, and identical residues in the other ADHs are marked with dashes. Secondary structure elements of β-strand (bbbbb) and α-helix (aaaaa) are shown below the aligned sequences.
Examination of the crystal structures of TbADH,31 CbADH,31 and EhADH129 at the respective resolutions of 2.5, 2.05, and 1.9 Å31, 32 revealed that all three ADHs are homotetramers comprising 38-kDa subunits sharing identical catalytic Zn binding sites.23, 28, 31 All three enzymes lack the four cysteines that in other bacterial tetrameric ADHs (e.g. Pseudomonas putida formaldehyde dehydrogenase, PFDH,33Sulfolobus sulfataricus ADH, SsADH34 and Pseudomonas aeruginosa ADH, PaADH,35 and dimeric eukaryotic ADHs like horse liver ADH, HLADH,36) form the structural Zn binding site. A comparison of the very similar crystal structures of holo-TbADH (PDB entry 1YKF) and holo-CbADH (PDB entry 1KEV) (r.m.s. difference in Cα = 0.8 Å) revealed several features that might account for the higher thermal stability of TbADH. One element that appears to be crucial is a proline substitution for Leu316 and Ser24, located at a β-turn and a terminating external loop in the polypeptide chain of CbADH (ΔT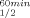 = 2.2 and 4.4°C, respectively).5
= 2.2 and 4.4°C, respectively).5
All the above findings provided the rationale for investigating the role of Pro100 in the thermal stability of all three enzymes.
Here, we report that a single substitution of the unique Pro100 found in EhADH1 for Gln100 in CbADH (Q100P-CbADH) significantly enhances the thermostability of the latter enzyme. We crystallized the mutant ADH and an analysis of its three-dimensional structure suggested that the proline substitution at this position stabilizes CbADH by reinforcing hydrophobic interactions and by reducing the flexibility of a loop at this strategic region.
MATERIALS AND METHODS
Site-Directed Mutagenesis
The enzymes for DNA cloning, sequencing, and amplification were purchased from Amersham (Buckinghamshire, England), New England Biolabs (Beverly, MA), Fermentas MBI (Vilnius, Lithuania), and Promega (Madison, WI). Oligonucleotides for cloning, sequencing, and site-directed mutagenesis of the T. brockii, E. histolytica, and C. beijerinckii adh genes were synthesized by the WIS Chemical Synthesis Laboratory. All other chemicals were of analytical grade. The genes encoding TbADH, CbADH, and EhADH1 were constructed and expressed according to Peretz et al.,28 using the plasmids pBSP80TbADH, pBSP80EhADH1, and pBSP80CbADH as templates for generating the recombinant plasmids coding for the respective mutants H100P-TbADH, P100A-EhADH1, and Q100P-CbADH. Site-directed mutagenesis of TbADH was performed according to the method of Byrappa et al.37 For EhADH1 and CbADH, we used the QuikChange XL Site-Directed Mutagenesis Kit of Stratagene (La Jolla, CA). The mutations were verified by DNA sequencing of the corresponding plasmids. The forward/reverse primers (5′-3′) used for generating the mutants are listed below, with the exchanged bases underlined:
for H100P-TbADH: CCCAGCACTCCGGTGGAA/GATATCCTCTTTGTACTTCAGA
for P100A-EhADH1: CGCAAAGAGGATATGCAATGCATTCAGGAGG/
CCTCCTGAATGCATTGCATATCCTCTTTGCG
for Q100P-CbADH: GTTCAAGCTGGTTTTCCACAGCACTCAAACGGTATGC/
GCATACCGTTTGAGTGCTGTGGAAAACCAGCTTGAAC
Expression and Purification of Recombinant Enzymes
All recombinant plasmids bearing TbADH, EhADH1, CbADH and their mutated variants were transformed into E. coli strain TG-1, and the recombinant proteins were purified according to a modification of a procedure described by Peretz and Burstein.26 Briefly, the transformed cells were cultured for 17 h in 2YT with zinc chloride (50 μM) and ampicillin (100 mg L−1) at 37°C, harvested by centrifugation for 20 min at 8,000g (4°C), and resuspended in buffer A (25 mM Tris-HCl, pH 7.3; 0.1 mM DTT, 0.1 mM EDTA, 0.1 mM benzamidine, 0.02% sodium azide, and 10% glycerol). The cells were disrupted for 5 min by pulsed sonication (Branson Sonifier 450) using a rosette cup immersed in an ice bath and then centrifuged (23,000g for 15 min) to remove cell debris. The supernatant was then heat-treated for 3 min at 65°C and recentrifuged for 15 min at 12,000g. The clear supernatant was applied onto a DEAE-52-cellulose column (10 × 5 cm2), pre-equilibrated with buffer A at 4°C. The column was extensively washed with buffer A until the eluate contained no detectable protein according to the method of Bradford.38 The proteins were eluted from the column with buffer A containing 0.1M NaCl, and the enzymatically active fractions were pooled and applied onto a short Red Sepharose column (12 × 5 cm2) (Pharmacia, Uppsala, Sweden). The purified recombinant enzymes eluted from the Red Sepharose column in a linear gradient of NaCl at concentration ranges of 0.1–1 M for TbADH, 0.1–2 M for CbADH, and 50–250 mM for EhADH1 in buffer A. The fractions containing alcohol dehydrogenase (ADH) activity were collected, concentrated by ultrafiltration (Amicon YM-30), and stored at 4°C. All mutant enzymes used in this study were obtained as intact tetramers and purified to homogeneity, as judged by Coomassie Brilliant Blue staining of SDS-polyacrylamide gel electrophoresis (PAGE) gels.
Enzyme Activity Assay
The catalytic activity of ADH at 40°C was measured by following the reduction of NADP+ (and the formation of NADPH), monitored at 340 nm (ε340 = 6.2 mM−1 cm−1). The standard assay mixture contained 150 mM 2-butanol, 0.5 mM NADP+, and 100 mM Tris-HCl (pH 9.0) in a total volume of 1 mL. One unit of ADH is defined as the amount of enzyme that catalyzes the oxidation of 1 μmol of 2-butanol min−1 at 40°C under the initial velocity at the above-mentioned conditions. Kinetic parameters were measured and calculated using a Beckman DU-7500 spectrophotometer equipped with a Multicomponent/SCA/Kinetics Plus software package and a thermostatted circulating water bath. The Km values for 2-butanol were determined using increasing concentrations of alcohol (0.1–100 mM) and enzyme (5–120 nM) with 0.5 mM NADP+ in 100 mM Tris-HCl (pH 9.0). Values represent the average of three experiments; individual measurements were within 10% of the quoted mean.
Thermal Inactivation (T1/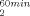 ; T1/
; T1/ )
)
The thermal stability of the ADHs was determined by monitoring the residual enzymatic activity after 60 min incubation in 25 mM Na/K phosphate buffer (pH 6.8) and 0.1 M NaCl at increasing temperatures. T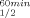 is the temperature at which 50% of the enzymatic activity is lost after 1-h incubation, interpolated from a plot of the residual enzymatic activity versus temperature. T
is the temperature at which 50% of the enzymatic activity is lost after 1-h incubation, interpolated from a plot of the residual enzymatic activity versus temperature. T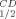 is the temperature at which 50% of the original CD signal at 218 nm is lost upon heating the protein sample between 30° and 98°C, with an average increase in temperature of 1°C min−1. The value is the mean of three experiments; individual measurements were within 5% of the quoted mean.
is the temperature at which 50% of the original CD signal at 218 nm is lost upon heating the protein sample between 30° and 98°C, with an average increase in temperature of 1°C min−1. The value is the mean of three experiments; individual measurements were within 5% of the quoted mean.
Circular Dichroism
Far-UV (200–260 nm) CD spectra were obtained at a temperature range of 30–110°C with an average increase in temperature of 1°C min−1, using an Aviv spectrophotometer, Model 202. Scans were performed every 4°C using ∼3 μM (0.5 mg mL−1) protein in 50 mM Na/K phosphate buffer, pH 6.8, in quartz cells and with a light path of 0.1 cm. Data (mdeg vs. wavelength) were collected every nanometer with an averaging time of 2 s. The background CD signal for each analysis, determined in a single scan using buffer alone at 25°C, was subtracted from each scan and was temperature-independent. The decrease in CD signal with increase of temperature was evaluated at the local minima of 218 nm and further normalized by subtracting from each wavelength the lowest signal observed at that particular wavelength. The change in the percent residual CD signal was plotted as a function of temperature, and the residual 50% CD signal (T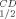 ) was estimated.
) was estimated.
Analytical Procedures
DNA sequencing was performed on an Applied Biosystems Model 373A DNA sequencer using the dideoxy method according to Sanger and Coulson39 and appropriate primers. SDS-PAGE was performed on 12% slab gels and 5% stacking gels according to Laemmli40 (Bio-Rad MiniProtean II system) and stained with Coomassie Brilliant Blue. Protein concentrations were determined according to Bradford,38 using bovine serum albumin as standard. The estimated molecular weight of the recombinant enzymes was determined by chromatography on a column of Superdex S-200.
Crystallization, Data Collection, and Refinement
Single crystals of the Q100P-CbADH mutant were obtained by the microbatch method under oil, using the IMPAX 1-5 robot (Douglas Instruments, East Garston, Hungerford, Berkshire, U.K.). The purified CbADH (6 mg mL−1) was crystallized in a mixture containing polyethylene glycol (PEG) 100 5% (w/v); 2-morpholinoethane-sulfonic acid (MES) 1M, pH 6; PEG 600 30% (v/v); glycerol 10% (v/v); 25 mM NaCl; 50 μM DTT and 25 μM ZnCl2. A complete data set was collected from a single crystal on a Rigaku R-AXIS IV++ imaging plate area detector using a Rigaku RU-H3R rotating anode operated at 5 kW and Osmic multilayer X-ray focusing mirrors.
Diffraction data were integrated, scaled, and reduced using the HKL program package.41 Crystals formed in space group P212121, with cell constants a = 79.47 Å, b = 103.34 Å, and c = 193.31 Å, contained one tetramer in the asymmetric unit cell with a Vm of 2.6 Å3 D−1 and diffracted to 2.25 Å resolution. The structure was solved by molecular replacement using the program PHASER,42 using the refined structure of an apo-CbADH mutant with enhanced thermostability30 (PDB accession code 1JQB) as a model. To avoid model bias in the mutated regions, we calculated a simulated annealing omit electron density map using the standard CNS protocol in a 3.5 Å vicinity of the mutated residues. Side chains of the mutated amino acid residues were then built into the resulting 3Fo-2Fc map. The data collection and refinement statistics are presented in Table I.
| Data collection | |
|---|---|
| Space group | P212121 |
| Unit cell dimensions | |
| a (Å) | 79.47 |
| b (Å) | 103.34 |
| c (Å) | 193.01 |
| Number of molecules in the asymmetric unit | 4 |
| Resolution range (Å) | 40–2.25 (2.29–2.25) |
| Number of reflections measured | 859,533 |
| Number of unique reflections | 75,865 (3760) |
| Rsym | 0.095 (0.373) |
| Completeness (%) | 99.2 (100) |
| Redundancy | 7 |
| 〈I〉/〈σ(I)〉 | 18 (3) |
| Refinement statistics | |
| Resolution limits (Å) | 40–2.25 (2.39–2.25) |
| Rfree | 23.0 (25.8) |
| Rwork | 19.3 (21.5) |
| Total number of reflections | 75,708 |
| Number of non-H-protein atoms | 11,080 |
| Number of water molecules | 590 |
| Mean B factor (Å2) | 30.8 |
| RMSDs from ideal values | |
| Bond lengths (Å) | 0.006 |
| Bond angels (°) | 1.3 |
| Torsion angles (°) | 23.4 |
| Improper torsion angles (°) | 0.76 |
| Estimated coordinate error | |
| Low resolution cutoff (Å) | 5 |
| ESD from Luzzati plot (Å)d | 0.24 |
| ESD from SIGMAA (Å) | 0.16 |
| Ramachandran statistics | |
| Most favored (%) | 87.9 |
| Additional allowed (%) | 11.4 |
| Generously allowed (%) | 0.3 |
| Disallowed regions (%) | 0.3 |
- Rsym = Σ|〈Ihkl〉 − Ihkl|/|Ihkl|, where 〈Ihkl〉 is the average intensity over symmetry-related reflections and Ihkl is the observed intensity. R = Σ‖Fo | − |Fc‖/Σ|Fo|, where Fo denotes the observed structure factor amplitude and Fc the structure factor calculated from the model. Rfree is for 10% of randomly chosen reflections excluded from the refinement.
All steps of atomic refinement were carried out using the program CNS.43 Map display and model rebuilding were performed using the program O,44 and model inspection using WHATIF and PROCHECK.45 The PDB accession code for Q100P-CbADH is 2B83. The figures were created using the program PyMOL (DeLano Scientific LLC).
RESULTS
Structural Considerations
The structure alignment of the monomers of CbADH, TbADH, and EhADH1 showed very high homology with rms difference of 0.8 Å between CbADH and EhADH1 and 0.7 Å between TbADH and EhADH1. A detailed analysis of the regions in close proximity of Gln100 in CbADH and His100 in TbADH showed that the phi and psi angles of Q100 of CbADH (−55, −41) and H100 of TbADH (−63, −22) fall into the zone allowed for prolines (ϕ −50 to −80; ψ = 120–180) or (ϕ −50 to −70; ψ = −10 to −50).15 Moreover, the preceding residues F99 of CbADH and Y99 of TbADH have the respective ϕ and ψ angles (−128, 75) and (−118, 77), the most common conformation for a residue preceding proline.46 The mutations Q100P-CbADH and H100P-TbADH were performed “in silico” and all possible Pro rotamers were examined. None of the newly introduced Pro rotamers clashed with other residues of the mutant protein.
Molecular and Enzymatic Properties of Mutant Enzymes
Following the site-directed mutagenesis of residue 100 in all three enzymes, we constructed the mutants Q100P-CbADH, H100P-TbADH, and P100A-EhADH1. The purified recombinants were characterized by non-denaturing gel filtration (Superdex S-200) and SDS-PAGE displaying the apparent molecular weights of 38 kDa per monomer and 160 kDa per tetramer, similar to those of the wild-type enzymes (data not shown).
Table II shows the kinetic properties of the enzymatic activity of TbADH, EhADH1, CbADH and their mutants at 40°C, using 2-butanol as substrate. The Km and the enzymatic efficiency (Kcat/Km) values of Q100P-CbADH were very similar to those of CbADH, indicating that neither the binding nor the catalytic efficiency of the enzyme was severely affected by the mutations. The Kcat of P100A-EhADH1 was reduced roughly twofold as compared to the wild type enzyme, however it was compensated by the subsequent reduction in Km leading to preservation of the enzymatic efficiency. Indeed, the mutation of residue 100 in the ADH is far from the active site of the enzyme,31 and any structural change that might have resulted from these mutations did not appear to perturb the active site. The enzymatic efficiency of H100P-TbADH was reduced about fourfold compared to the native enzyme, mainly due to the reduced substrate binding (three fold elevation in Km). This result supports the notion that a conformational change caused by a mutation located at one end of a protein molecule may induce a conformational change at another end of the molecule, thus affecting properties of the protein.47
| ADHs | Km (mM) | Kcat (min−1) | Kcat/Km (min−1 mM−1) |
|---|---|---|---|
| TbADH | 3.1 | 1724 | 556 |
| H100P-TbADH | 10.2 | 1250 | 125 |
| EhADH1 | 0.74 | 3025 | 4088 |
| P100A-EhADH1 | 0.3 | 1588 | 5295 |
| CbADH | 13 | 9714 | 747 |
| Q100P-CbADH | 14 | 9142 | 653 |
- Enzymatic activity was measured by monitoring the formation of NADPH at 340 nm. Km values for 2-butanol were determined with varying concentration of 2-butanol (0.1–100 mM), enzyme (5–120 nM), and 0.5 mM NADP+ in 100 mM Tris-HCl (pH 9) in a total volume of 1 ml, in triplicates. Values are the averages of two experiments and the individual measurements were within 10% of the quoted mean. ADHs, Alcohol dehydrogenase.
Thermal Stability
The T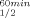 and T
and T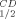 values of the mutants are presented in Table III and in Figures 2 and 3, respectively. Replacing Gln100 with Pro significantly enhanced the thermal stability of CbADH: ΔT
values of the mutants are presented in Table III and in Figures 2 and 3, respectively. Replacing Gln100 with Pro significantly enhanced the thermal stability of CbADH: ΔT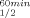 = +8°C; ΔT
= +8°C; ΔT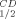 = +11.5°C. Replacing His100 of TbADH with Pro and Pro100 of EhADH1 with Ala, however, did not alter the thermostability of the thermophilic enzymes.
= +11.5°C. Replacing His100 of TbADH with Pro and Pro100 of EhADH1 with Ala, however, did not alter the thermostability of the thermophilic enzymes.

The effect of the mutations on the thermal stability of the enzymatic activity of the ADHs. The thermal stability of the enzymes was determined by monitoring their residual enzymatic activity after 60 min incubation at varying temperatures. Enzymatic activity was assayed by spectrophotometric analysis of NADPH formation at 340 nm (as described under materials and methods). Values are expressed as the averages of at least three experiments.

The effect of the mutations on the thermal stability of the secondary structure of the ADHs. Thermal stability of the enzymes was determined by monitoring the loss of their CD signal at 218 nm upon heating. The data (mdeg vs. wavelength) were collected every nm with an averaging time of 2 s using quartz cells with a light path of 0.1 cm. Experiments were performed between 30°C and 98°C, with an average increase in temperature of 1°C/min. Scans were performed every 4°C. The protein concentration was about 0.5 mg mL−1 in 50 mM Na/K phosphate buffer (pH 6.8). The background for each scan was determined in a single scan with buffer alone at 25°C and subtracted from each scan. The background CD signal was temperature-independent.
| ADHs | T (°C)a (°C)a |
ΔT (°C)b (°C)b |
T (°C)a (°C)a |
ΔT (°C)b (°C)b |
|---|---|---|---|---|
| TbADH | 93.8 | 93.8 | ||
| H100P-TbADH | 93.8 | 0 | 93.8 | 0 |
| EhADH1 | 77.5 | 80 | ||
| P100A-EhADH1 | 77.5 | 0 | 78.5 | −1.5 |
| CbADH | 64.5 | 65.5 | ||
| Q100P-CbADH | 72.5 | +8 | 77 | +11.5 |
- ADHs, Alcohol dehydrogenase.
- a
T
 is the temperature at which 50% of the enzymatic activity is lost after 1 h incubation, interpolated from a plot of the residual enzymatic activity vs temperature. T
is the temperature at which 50% of the enzymatic activity is lost after 1 h incubation, interpolated from a plot of the residual enzymatic activity vs temperature. T is the temperature at which 50% of the original CD signal at 218 nm is lost upon heating the protein sample between 30 and 98°C, with an average increase in temperature at 1°C/min.
is the temperature at which 50% of the original CD signal at 218 nm is lost upon heating the protein sample between 30 and 98°C, with an average increase in temperature at 1°C/min.
- b Relative to the wild type enzyme.
Three-Dimensional Structure of Q100P-CbADH
The three-dimensional structure of Q100P-CbADH [tetramer, Fig. 4(a)] was determined to 2.25 Å resolution. No significant change was observed in the Cα chain of the mutant compared with that of the wild-type protein. Inspection of the three-dimensional structure of Q100P-CbADH shows that substitution of the side chain of glutamine with the pyrolidine ring of proline did not impose any steric constraint on the protein structure. Two intra-molecular hydrogen bonds were lost, one between Oϵ1 of the amide group of Gln100 and the main chain nitrogen of Gly297 (distanced 2.62 Å), and the other between Nε2 of the amide group of Gln100 and the main chain carbonyl of Gly297 (distanced 2.92 Å). Upon mutation, an extra CH2 group, Cδ of Pro100, was introduced into a non-polar environment formed by the nearby Pro88 (distant 3.8 Å from the mutation site), Trp90 (3.5 Å from the mutation site), and Val95 (4 Å from the mutation site) [Fig. 4(b)]. All these residues—P100, P88, W90, and V95—are located in the structural lobe of the protein. An additional 11 aliphatic and aromatic carbon atoms are present within the sphere of 6 Å from Cδ of Pro100 (two methyl groups of Val95; three carbon atoms of the Trp90 indole group; Cβ and Cγ methylene groups of Pro100; Cβ and Cγ of Gln101, and two carbons of the Phe99 phenyl ring).

(a) Ribbon diagram of the CbADH tetramer. Subunits are represented in different colors. Gln100 is depicted in ball representation in all four subunits. (b) Superimposition of the structural lobes of CbADH (green) and the Q100P-CbADH mutant (cyan). The side chains of residues 88, 90, 95, and 100 are indicated. (c) View of the inter-subunit interactions in EhADH1 by the seven member ion-pair network: R97A–E93A–K257B–D237B–K304C–D168B–K238B and of the R97A–P258B and R97A–G259B charged neutral hydrogen bonds (the superscript refers to the subunit). The ion pairs and the hydrogen bond lengths are shown in Å.
DISCUSSION
The stabilization of proteins by reducing the entropy of the unfolded state through the introduction of proline residues is well documented.5, 14, 17 Proline seems an ideal candidate for thermostabilizing proteins because its pyrrolidine ring adopts fewer conformations than does any other amino acid. Watanabe19, 20 analyzed the critical sites for protein thermostabilization by proline. According to the ‘Suzuki rule’, a protein can be stabilized when prolines are inserted at certain strategic positions—namely, the second site of the beta turn or the N-cap of the alpha helix and flexible loops. The authors also suggested that the contribution of each proline mutant influences the stability of the protein in an independent and an additive manner.
Several reports support the Suzuki theory. For example, xylose isomerase from Thermoanaerobacterium thermosulfurigenes was thermostabilized by the substitution of a proline residue for Gln58 (within a large loop) but for example not for Ala62.17 The thermostability of barley (1→3,1→4)-β-glucan endohydrolase was increased when His300 was changed to Pro (in a terminal loop).18 Muslin et al. stabilized barley α-glucosidase by replacing its N-cap Thr340 residue with Pro.16
This strategy proved to be very successful in the current study. The significant increase of T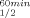 gained by the substitution of Pro for Gln100 in CbADH (ΔT
gained by the substitution of Pro for Gln100 in CbADH (ΔT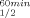 = +8°C) is approximately up to 4-fold higher than that achieved by substitution with any of the non-conserved prolines of TbADH.5
= +8°C) is approximately up to 4-fold higher than that achieved by substitution with any of the non-conserved prolines of TbADH.5
Residue 100 of CbADH is part of the protruding lobe of the protein comprising residues 87–110 and represents a loop connecting strands β5 and β6, and engulfing a short 5-residue α-helix, α2 [residues 93–97, Fig. 1]. The lobe is a conserved structural feature within the medium chain dehydrogenase/reductase (MDR) super family.31, 34, 36, 48 In the three enzymes studied here, this structural lobe plays a crucial role in maintaining the quaternary structure of the enzymes. The basic tetrameric structure of the ADHs in all three enzymes is a dimer of dimers, in which subunits A and B (or C and D) form the main horse liver-ADH-like dimer. Two such dimers (AB and CD) associate through subunits A and C (or B and D), and A and D (or B and C) to form the tetramer [Fig. 4(a)].31 Many of the inter-subunit interactions that facilitate the formation of the tetramers of CbADH,31 EhADH1,32 and TbADH31 are maintained by the protruding lobe of the catalytic domain located at the dimer-dimer interface.
One explanation for the dramatic impact exerted by the Gln to Pro mutation at position 100 on the thermostability of CbADH, as opposed to the negligible effect observed in TbADH and EhADH1 upon mutation of residue 100, could be that the lobe of the mesophilic CbADH is much less confined than that of the thermophilic ADHs. In the extreme thermophile, TbADH, the lobe is stabilized by side-chain to main-chain inter-subunit ‘charge-neutral’ hydrogen bonds that are absent in the mesophilic CbADH: Arg97A–Pro258B and the three other identical hydrogen bonds, related by protein 222 symmetry.31 Similarly the structural lobe of the thermophilic EhADH1 participates in several inter-subunit interactions that are not found in the mesophilic CbADH: Arg97 and Glu93 of the lobe participate in a seven-residue inter-subunit ion pair network: Arg97A–Glu93A–Lys257B–Asp237B–Lys304C–Asp168B–Lys238B. This lobe also forms a ‘charge-neutral’ intersubunit hydrogen bond of Arg97A–Pro258B and Arg97A–Gly259B [Fig. 4(c)].32 We propose that the stabilization of the protruding lobe of Q100P-CbADH is due to the reduction of the entropy of the unfolded state because (a) proline can adopt only a limited number of conformations as compared with other amino acids and (b) proline restricts the conformation of the presiding residue.14, 46
Replacing the polar Gln100 with a non-polar proline within a vicinity of three other hydrophobic residues of the protruding lobe [W90, P88 and V95; Fig. 4(b)] increased the hydrophobic interactions in this region (the estimated hydrophobic effect for residue burial is 3.1 kcal/mol for Pro and 1.65 kcal/mol for Gln).49 Fersht and coworkers showed a linear correlation between the free energy change of a protein and the number of methyl and methylene groups located within a 6 Å-radius of deleted hydrophobic groups.50, 51 Deletion of a single methylene group from a non-polar environment of 10 other methylene and methyl groups located within a 6 Å distance of the mutation yielded a destabilization effect of ca 0.5 kcal mol−1.51 Similarly, introduction of the Cδ of Pro100 into a nonpolar environment of the protruding lobe of Q100P-CbADH could have contributed to the extensive stabilizing effect of this mutation. This type of stabilization resembles the stabilization effect that occurs in structural Zn-containing MDRs (including the thermophilic Sulfolobus, and the Aeropyrum sp. ADHs and the horse liver ADH),34, 36, 48 in which the protruding lobe is stabilized by four cysteine residues placed on both sides of the lobe—Cys 97 (or Glu in the Sulfolobus sp. and Asp in Aeropyrum pernix), Cys 100, Cys 103, and Cys 111 (HLADH numbering), ligating to a single structural Zn ion placed in the center of the lobe. In these enzymes, Zn is essential for stability.36, 52 Recently Watanabe et al. have demonstrated that the thermal stability of the xylitol dehydrogenase from the mesophilic fungi Pichia stipitis was increased due to insertion of structural zinc at the lobe of the enzyme.53 It seems therefore that within the MDR super family the structural lobe may serve as a target for stabilization, by strategically inserted proline residues.
Acknowledgements
We thank Prof. F. Frolow and Dr. L. Shimon for helpful discussions and Dr. Virginia Buchner for helpful remarks during manuscript preparation. The three-dimensional structure of Q100P-CbADH was solved at the Israel Structural Proteomics Center (ISPC).

