

Melting of solvents nanoconfined by polymers and networks
Abstract
Thermoporosimetry is becoming increasingly used to study nanoscale heterogeneity and structure in polymer networks. The starting point for thermoporosimetry is the Gibbs-Thomson (GT) relation between melting point and inverse crystal size. In the case of polymers, the Flory-Huggins (FH) model also predicts that there is a depression of the melting point because of the mixing of the polymer and the solvent molecules, and this needs to be taken into account. The first step in analysis of size heterogeneity using thermoporosimetry and the GT equation requires that there be quantitative agreement between the FH theory and the melting points of the diluent in the uncrosslinked rubber. We find that both benzene and hexadecane exhibit excessive melting point depressions in uncrosslinked polyisoprene. This may imply that the uncrosslinked polymer is divided into ‘nanoheterogeneities’. We further find that the heat of fusion decreases as polymer concentration increases for the benzene, but not for the hexadecane. Finally, we compare pore size distributions obtained for a crosslinked polyisoprene as determined from the melting behavior of n-hexadecane as a diluent, using different references for how the uncrosslinked polymer behaves. While number average distribution is not very different between the different analyses, the weight average distribution is. © 2006 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 44: 3475–3486, 2006
BACKGROUND AND INTRODUCTION
Heterogeneity is important in glass forming materials and one potential means of controlling heterogeneity at the nanoscale is to change crosslinking density of rubber networks. There are two ways in which to estimate network structure from polymer-diluent interactions. The first is to use Flory-Rehner swelling theory1 to estimate an average molecular weight between crosslinks, but this method provides only a mean value and has additional quantitative issues such as the crosslink dependence of the mixing thermodynamics.2-6 We do not consider this method here. The method of thermoporosimetry has come into some use as a means of characterizing heterogeneous systems and crosslinked polymer heterogeneity.7-13 In this method, thermal analysis is used to obtain melting point distributions and, through the Gibbs-Thomson (GT) equation,14 relate crystal size and size distribution to the shape of the melting thermograms. In the case of crosslinked rubber, the melting point depression has an additional thermodynamic contribution different from size that is related to the polymer–solvent interactions and should be described by the Flory-Huggins (FH) expression15, 16 for melting of the diluent in a polymer solution.
The present work stems from our plan to characterize dynamics of glass forming liquids constrained in rubber networks crosslinked to different degrees. Thermoporosimetry was to be used to characterize the heterogeneity. As seen in what follows, one of the major assumptions of thermoporosimetry in such systems that the FH thermodynamics describes the melting of small molecule diluents in uncross-linked polymers is not valid and furthermore is not readily “fixed” by a change in the mixing thermodynamics by a small change in the FH interaction parameter χ. We explore the consequences of this finding in the Discussion section.
The article is laid out as follows. After the introductory section, we present the equations for melting point depression from both FH theory and from the so-called GT equation. This is followed by a description of our experimental methods and a section on results for benzene and n-hexadecane in uncrosslinked polyisoprene and the relevant phase diagrams. We then present a discussion of the results in terms of finite size effects based on the melting of selected diluents in controlled pore glasses. We also show the differences for the estimated size distribution based on thermoporosimetry analysis that accounts for deviations from FH and that which does not. Finally, we provide a set of conclusions.
Thermodynamic Analysis
Melting and Thermodynamics of Mixing
 (1)
(1) (2)
(2)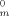 are the melting temperatures in the solution and in the bulk and ΔHu and Vu refer to the heat of fusion and volume per mole repeating unit, respectively. v refers to the volume fraction, x is the degree of polymerization of the polymer, and χ is the polymer–diluent interaction parameter. At equilibrium between liquid and crystalline diluent in the polymer solution, the chemical potentials of the diluent in the two phases must be equal. At the melting temperature of the pure diluent, its chemical potential equates to that in the standard state.17 Importantly, the melting point depression of the diluent occurs upon mixing with the polymer because the chemical potential of the diluent in solution is less than that of the pure solvent. This affects the melting of the solvent in that system.
are the melting temperatures in the solution and in the bulk and ΔHu and Vu refer to the heat of fusion and volume per mole repeating unit, respectively. v refers to the volume fraction, x is the degree of polymerization of the polymer, and χ is the polymer–diluent interaction parameter. At equilibrium between liquid and crystalline diluent in the polymer solution, the chemical potentials of the diluent in the two phases must be equal. At the melting temperature of the pure diluent, its chemical potential equates to that in the standard state.17 Importantly, the melting point depression of the diluent occurs upon mixing with the polymer because the chemical potential of the diluent in solution is less than that of the pure solvent. This affects the melting of the solvent in that system.Melting and Finite Size Effects
 (3)
(3)EXPERIMENTAL
Materials
The solvents used in this study are benzene and n-hexadecane, both of which are of GC grade and obtained from Sigma-Aldrich, Co. without further treatment. cis-1,4-polyisoprene was obtained from Polysciences with weight average molecular weight of 305,000 g/mol and polydispersity of 1.05. Polyisoprene was crosslinked by dicumyl peroxide (DCP) at 149 °C for 2 h, which results in virtually complete reaction of the peroxide. The crosslink density was controlled by varying the amount of added dicumyl peroxide, which was represented as parts of DCP per hundred parts of polyisoprene (phr). Samples are designated phr1, phr5, and phr 10 for DCP quantities of 1, 5, and 10 phr, respectively. DCP is considered a quantitative crosslinker for polyisoprene,18 and the average molecular weight between crosslinks is estimated to be 13.95, 2.76, and 1.36 kg/mol for the phr1, phr5, and phr10 crosslinked polyisoprene, respectively.19
To calibrate the relationship between melting point depression and pore size, controlled pore glasses (CPGs) from Millipore with known mean pore size and narrow pore size distribution were used. The mean pore diameter of the CPGs ranged from 8 to 110 nm. The CPGs were cleaned and surface treated following the procedures in ref.20 to promote wetting by organic liquids.
Methods
The melting behavior of solvents in uncrosslinked and crosslinked polyisoprene was studied using a Perkin–Elmer Pyris 1 differential scanning calorimeter (DSC). Polymer and solvent were weighed separately into hermetic pans, and the sealed pans were kept at room temperature for at least 24 h before experiments were carried out to obtain a homogeneous solution. Pan weights were checked during this time as well as before and after experimental runs to ensure no solvent leakage, which is especially important for the benzene-polyisoprene mixtures. For n-hexadecane in controlled pore glasses, DSC samples were prepared in the same way except that hermetically sealed pans were heated at 100 °C for an additional 3 h to assure the filling of the nanopores with the n-hexadecane.
The temperature and heat flow of the DSC were calibrated using standard references. A cooling scan was first run at 5 K/min to −50 °C and the instrument held isothermally at −50 °C for 15 min. This was followed by a heating scan at 5 K/min. Melting temperatures were determined from the onset melting point during heating runs.
For the glass-transition temperature measurements of the benzene-polyisoprene mixtures, a cooling scan at 20 K/min down to −140 °C was first performed, which was immediately followed by a heating scan at 10 K/min to obtain the limiting fictive temperature, which is equivalent to glass-transition temperature.21
To obtain the “pore” size distribution of crosslinked polyisoprene, the calibration of “pore” diameter was performed using the CPGs with known pore size and narrow pore size distribution. By means of the Gibbs-Thomson (GT) equation, the melting point depression of confined n-hexadecane in CPG was related to the size of confinement, which is the “pore” size of the CPG. After determining the relationship between melting point depression and the “pore” size of the confinement, the “pore” size distribution of the crosslinked polyisoprene was obtained from the melting thermograms of the polyisoprene-confined n-hexadecane.
 (4)
(4)RESULTS
Solvent Melting in Uncrosslinked Polyisoprene
The DSC thermograms for the benzene/polyisoprene system at different concentrations are shown in Figure 1. As the volume fraction of polyisoprene increases, the melting endotherm of the benzene shifts to lower temperatures and broadens. The results for the melting temperature and the heat of fusion of benzene in the uncrosslinked polyisoprene are shown in Figure 2. Figure 2(a) also includes the FH prediction for melting point depression. A concentration independent FH parameter of χ = 0.4319 was used for the theoretical estimation. If peak melting temperature is used to determine ΔTm, the melting point depression is close to, but still greater than the estimation based on FH theory. If onset melting temperature is chosen, however, one can see a large deviation from the FH theory in Figure 2(a). Here, because of the broad endotherm and our interest in deviations from FH theory, we use the onset melting point, rather than the just the peak maximum, to characterize the melting behavior of the diluent. We remark that when the melting endotherm is narrow, there is little difference between the two. Furthermore, it is a common practice to use the peak melting to describe size dependence of the melting behavior.19, 20, 22-24 In such cases, the interest has been in the general trends and not in the details of broadening, per se. In the case of thermoporosimetry, the entire thermogram is of interest and one can see from the above discussion that the peak is related most closely to the weight average pore diameter while the onset emphasizes the smallest crystals (or other event in the case of deviations from FH theory). As shown in Figure 2(b), there is a decrease in the heat of fusion of benzene at high polymer concentrations. To further examine this issue, plasticization effects due to noncrystallizing benzene were studied by investigating the glass-transition temperature of the benzene/polyisoprene mixture. The results indicate that there is some plasticization caused by the benzene at high polymer concentrations, but this accounts only partially for the decrease in the heat of fusion. This is discussed further subsequently.

DSC thermograms of benzene in uncrosslinked polyisoprene at different volume fraction of polymer (v2).

Benzene/polyisoprene system: (a) melting point depression. The dashed line refers to a least-squares regression of exponential growth with correlation coefficient of 0.996; (b) the heat of fusion of benzene. The line through data represents a guide for the eye, and the bottom three data points at high polymer volume fraction have not been corrected by the plasticization effect. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
We also examined n-hexadecane as the diluent in the polyisoprene. Its DSC thermograms and melting behavior in the presence of uncrosslinked polyisoprene are shown in Figures 3 and 4, where again we show melting point depression as determined by onset value. Once again melting point depression does not follow the FH prediction and the deviation depends on the volume fraction of polyisoprene v2. As v2 increases, the deviations from the FH estimation increase. In terms of the heat of fusion, the n-hexadecane displays a quite different behavior from the benzene. Instead of decreasing, the heat of fusion for the n-hexadecane remains almost constant over the full concentration range investigated. This observation satisfies the assumption underlying FH theory, which assumes that the heat of fusion in the solution is equal to that in the bulk. In spite of this, the n-hexadecane/polyisoprene system still exhibits an evident deviation from the FH prediction for the melting point depression.

The DSC thermograms of n-hexadecane in the presence of uncrosslinked polyisoprene (v2 is the volume fraction of polyisoprene). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]

n-hexadecane/polyisoprene system: (a) onset melting point depression. The dashed line is a least-squares regression of exponential growth with correlation coefficient of 0.997; (b) heat of fusion of n-hexadecane. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Here, because of the unavailability of a literature value for the χ parameter for the n-hexadecane and polyisoprene mixture, we took χ = 0.5, which is reasonable for n-alkane and polyisoprene systems.25 Although we did not take into account the possible dependence of χ on concentration, it is not expected to have a great impact here.
A common method of using the FH theory to obtain an “empirical” fit to the behavior of polymer solutions is to make the χ parameter an adjustable parameter.26 If we do this here, we find that for the n-hexadecane, an unreasonable value for the interaction parameter (χ ≈ −0.4) would be required. Similarly, in the case of the benzene/polyisoprene system, to make the onset melting point consistent with the FH prediction, the FH interaction parameter χ needs to be zero (χ ≈ 0), which is not reasonable for polymer–diluent systems.
“Phase” Diagrams
Considering the complex melting behavior, especially the decrease in the heat of fusion shown by the benzene/polyisoprene system, we constructed its “phase” diagram as shown in Figure 5. The diagram includes the nonequilibrium glassy and supercooled liquid states in addition to the true thermodynamic states. In Figure 5, the squares represent glass-transition temperature data for benzene/polyisoprene solutions, which is accounted for by the appreciable plasticization effect from the vitrified benzene/polyisoprene “supercooled” liquid phase in the high polymer concentration regime. The dotted line in the lower part of “phase” diagram is the prediction for Tg of the mixtures based on the empirical Fox equation.28 As can be seen, at a volume fraction of polymer less than around 0.6, the experimentally observed Tg data depart significantly from the Fox relation. In fact, the Tg from v2 ≈ 0.17 to v2 ≈ 0.53 is almost constant and consistent with a volume fraction benzene of about 10% being dissolved in the polyisoprene. Importantly, the phase diagram glass transition data do not show inconsistencies with the literature29, 30 because we do not see, e.g., two glass transitions in our system, perhaps because of the difficulty of glass formation in benzene. The left side of the phase diagram in Figure 5 shows a surprising jump in the Tg of the system at a composition of around 53% polymer, i.e., we see that the jump is to a Tg that is very close to the bulk Tg of polyisoprene indicating that at concentrations lower than about 53% polymer, the benzene phase separates nearly completely and, therefore, the Tg observed is that of the polyisoprene rather than of plasticized polyisoprene. In contrast, at the higher polymer concentrations or, perhaps, in the small confinements, the glass forming ability of benzene improves. As a result above 53% polymer, as shown in Figure 5, the glass transition of the mixture exhibits evident composition dependence, which suggests the forming of the glassy or supercooled benzene/isoprene mixture.

“Phase” diagram of benzene/polyisoprene. Glass-transition temperature for pure benzene is from ref.27. The dashed line on the top represents the FH prediction for melting temperature of polyisoprene in the mixture based on eq 2; the dotted line at the bottom is the estimation for glass-transition temperature of the mixture based on the Fox equation and the horizontal dashed line refers to the theoretical prediction for the eutectic point in this system. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
We did not measure the melting of polyisoprene as it is a slow crystallizer, therefore our FH estimate for the melting of polyisoprene, shown by the dashed line in Figure 5, was based on literature data31 for the polyisoprene parameters required by eq 2. Since there is not a large difference between the melting temperatures of benzene and polyisoprene, it is theoretically expected to have a eutectic point32-35 at v2 ≈ 0.5, where the two solid phases of ployisoprene and benzene are simultaneously in equilibrium with the isotropic liquid phase. We present this in Figure 5 for completeness; in the hypoeutectic concentrations, there is crystallized benzene and supercooled polyisoprene/benzene in our system. Interestingly, it appears that this hypoeutectic system is a supercooled mixture of 10% benzene with polyisoprene plus pure benzene crystals. Above the eutectic composition, the benzene in the supercooled mixture takes on the composition of the glassy system, which now decreases from about 35% to 15% benzene in the hypereutectic range investigated. We comment that the details of this behavior could conceivably be affected by the kinetics of crystallization of the benzene during cooling.
For n-hexadecane and uncrosslinked polyisoprene, because of the nearly constant heat of fusion, plasticization is not expected, which makes its “phase” diagram much simpler than that for the benzene/polyisoprene system as illustrated in Figure 6.

Phase diagram of n-hexadecane/polyisoprene. Circles represent the measured onset melting temperature of n-hexadecane; the upward dashed line is the FH prediction for the melting temperature of polyisoprene; the horizontal dashed line refers to the eutectic line by theoretical estimation. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Solvents Nanoconfined in Controlled Pore Glasses
To investigate whether or not finite size effects provide an explanation for the observed breakdown of the FH prediction for diluent melting in uncrosslinked polyisoprene, we employed CPGs as “calibrants” for the diluent melting behaviors. The results for n-hexadecane in the CPGs are presented in Figure 7, which shows a linear relationship between melting point depression and inverse crystal size, as predicted by the GT eq 3. Assuming cylindrical pores, the surface energy of the solid–liquid interface was determined to be 13.9 erg/cm2, which is in reasonable agreement with literature values for other alkanes (σsl = 7.2 erg/cm2 for n-heptadecane and σsl = 9.6 erg/cm2 for n-octadecane36).

n-hexadecane in controlled pore glasses: (a) onset melting point depression. The solid line represents least-squares regression of data with the correlation coefficient of 0.998; (b) heat of fusion of n-hexadecane. The line through data is a guide for the eye. The inset shows the plot of the heat of fusion as a function of reciprocal of pore diameter, where the least-squares regression was represented by the straight line with the correlation coefficient of 0.996. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
 (5)
(5) (6)
(6)The melting point depression and the heat of fusion of benzene confined in the CPGs are also shown in Figure 8. Except for the smallest pore size studied (d = 8 nm), the melting point depression of the confined benzene follows a linear relationship with respect to reciprocal of the pore diameter. The heat of fusion also decreases with decreasing pore diameter. For the benzene in the uncrosslinked polyisoprene, we found that the crystal size estimated from melting point depression and heat of fusion calibration gave similar results, i.e., crystal sizes range from 8 to 40 nm.

Benzene in controlled pore glasses: (a) onset melting point depression. The solid line represents least-squares regression of data at d > 8 nm; (b) heat of fusion of benzene. The line through data is an exponential decay fitting with the correlation coefficient of 0.964. The inset is the plot of the heat of fusion against the reciprocal of pore diameter and the straight line represents the linear regression with the correlation coefficient of 0.962. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Both benzene and n-hexadecane exhibit melting point depressions and heat of fusion depressions upon confinement in controlled pore glasses. However, only benzene shows a depression of the heat of fusion in the uncrosslinked polyisoprene. Hence, while finite crystal size in the polyisoprene/benzene system may account for the deviations from the FH prediction, it appears that such an explanation is not consistent with the n-hexadecane results. It appears that another explanation for the breakdown of the FH model of diluent melting in uncrosslinked polymers needs to be found.
DISCUSSION
Beyond the fundamental finding that the FH expression for melting point depression of the small molecule diluent does not apply to the diluent/polyisoprene systems investigated here, the importance of the present work relates to the correct choice of the “baseline” to use in applying thermoporosimetry to characterize polymer networks. Should one use the FH theoretical baseline or should one use the empirically determined melting of the diluent in the uncrosslinked polymer? In our discussion, we consider the behavior of the diluents in the crosslinked polymer and the crystal-size analysis based on the ideas presented above using both FH and GT relationships and taking into consideration the findings of the breakdown of FH for the uncrosslinked polymer.
Figure 9 shows the melting point depression of n-hexadecane confined in crosslinked polyisoprene networks. It is found that ΔTm depends not only on the polymer concentration, but also on the crosslink density of the polyisoprene network. With larger crosslink density, which implies a smaller network “pore” size, the confined solvent displays a greater melting point depression. Figures 10(a,b) show the number average “pore” size distributions calculated from different “baselines”. Although the network “pore” size distributions for both methods are similar (mean diameter ≈ 4.0 nm) when using the number fraction, larger differences can be seen when the pore size distribution is expressed in terms of the weight fraction instead of the number fraction [Fig. 10(c)]. Figure 10(c) shows only the weight average results for the FH baseline because of the fact that at low values of ΔT, there is overlap between the empirical baseline and the measurements for the crosslinked system. Furthermore, it is worth reminding the reader that use of the “baseline1” (FH baseline) to obtain Figure 10(c) can be justified because the n-hexadecane/polyisoprene system does not behave as if the cause of failure of the FH theory in the uncrosslinked rubber is finite size effects, that is, we treat it as “intrinsic” (see above discussion as well).

Plot of onset melting point depression of confined n-hexadecane in polyisoprene networks. phr stands for part of DCP per hundred parts of polyisoprene, which represents crosslink density. The dashed line for crosslinked polyisoprene refers to a guide to the eye. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]

The number and weight average “pore” size distribution for phr10 polyisoprene network at v2 = 0.51 with different baselines. Based on n-hexadecane as the diluent, the size dependence of the heat of fusion obtained from CPG data was used for data analysis. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
We also consider the finding that the n-hexadecane/CPGs data shown in Figure 7(b) exhibits a significant pore size dependence of the heat of fusion, which was taken into account in generating Figure 10. But the heat of fusion of the n-hexadecane in the uncrosslinked polyisoprene was found to be nearly constant and independent of concentration. Hence, we replot the “pore” size distribution of the polyisoprene network by assuming a constant heat of fusion for the n-hexadecane in Figure 11 with solid bars. The weight average “pore” size distribution shows a slight difference from the case considering the size dependence of the heat of fusion [hollow bars in Fig. 11(c)], while the number average “pore” size distribution barely changes. Hence, a size-dependent heat of fusion does not seem to have a large effect on the estimated pore size distribution in this case.

The number and weight average “pore” size distribution for phr10 polyisoprene network at v2 = 0.51 with different baselines. Based on n-hexadecane as the diluent, both the constant heat of fusion and size dependent heat of fusion were used and compared for data analysis. The abscissa was arranged in the way corresponding to the same temperature interval. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Also, we determined the weight average “pore” size distribution using constant temperature (ΔT) intervals (rather than constant pore diameter intervals) to analyze the thermograms. This is reflected by the abscissa in Figure 11 and we see that the appearance of the weight-average pore size distribution looks like the thermograms obtained from DSC. The differences in the ways of analyzing data have potential significance for characterizing network heterogeneity in general as well as in the instance that originally motivated this work to use the network as a template for studying dynamics of glass-forming liquids in known heterogeneity environments. Unfortunately, one issue that arises in the thermoporosimetry is the fact that the origins of the FH breakdown are not known. For example, if it is because of finite size effects, the experimental melting curve also may not be able to be used as a baseline. If there is an “intrinsic” effect not related to finite size crystals in the uncrosslinked rubber, then it becomes possible to use the experimental melting curve for the diluent of interest in the uncrosslinked polymer.
SUMMARY AND CONCLUSIONS
By studying the melting behavior of benzene and n-hexadecane in uncrosslinked polyisoprene, we have found a breakdown of the FH prediction for the diluent melting point depression. An anomalous heat of fusion for the diluents is also found, which depends on both solvent type and polymer concentration. Although the heat of fusion of n-hexadecane is almost unchanged in solution with polyisoprene, which satisfies the assumption underlying FH theory, an unrealistic value of the FH interaction parameter would be required to bring its onset melting point depression into agreement with the FH prediction. In the case of crosslinked polyisoprene networks, the confined solvent exhibits an additional melting point depression compared with the uncrosslinked polymer and the depression is shown to be dependent on the crosslink density, which implies the finite size effect on melting temperatures and that the size depends on crosslink density.
To our knowledge, this is the first systematic work showing the anomalous melting point depression and a breakdown of FH predictions for the diluent melting in polymer–diluent systems. Beyond the fundamentals of the thermodynamics of melting in such systems, the significance of the present findings arises in thermoporosimetry where a correct contrast or “baseline” is needed to characterize the heterogeneity of crosslinked networks. It appears possible that thermoporosimetry should be carried out using a measured “baseline” of the melting temperature of the diluent in the uncrosslinked polymer, rather than the theoretical baseline from the FH theory, to calculate ΔTm. However, because the melting behavior of the hexadecane in polyisoprene is not fully consistent with this approach, further work needs to be performed. In particular, the hypothesis that melting point depression of diluent in uncrosslinked polymers (polyisoprene) is due to finite size effects suggests strongly that future work should undertake direct measurements of such effects. Small angle X-ray scattering or small angle neutron scattering to characterize actual crystal sizes and their distributions are possible routes for investigation.
Acknowledgements
This work was supported by the National Science Foundation under grant DMR-0304640.

