

FUN14 Domain-Containing 1–Mediated Mitophagy Suppresses Hepatocarcinogenesis by Inhibition of Inflammasome Activation in Mice
Abstract
Mitochondria lie at the heart of innate immunity, and aberrant mitochondrial activity contributes to immune activation and chronic inflammatory diseases, including liver cancers. Mitophagy is a selective process for removing dysfunctional mitochondria. The link between mitophagy and inflammation in tumorigenesis remains largely unexplored. We observed that FUN14 domain-containing 1 (FUNDC1), a previously characterized mitophagy receptor, accumulates in most human hepatocellular carcinomas (HCCs), and we thus explored the role of FUNDC1-mediated mitophagy in HCC initiation and progression in a mouse model in which HCC is induced by the chemical carcinogen, diethylnitrosamine (DEN). We showed that specific knockout of FUNDC1 in hepatocytes promotes the initiation and progression of DEN-induced HCC, whereas FUNDC1 transgenic hepatocytes protect against development of HCC. Hepatocyte-specific FUNDC1 ablation results in the accumulation of dysfunctional mitochondria and triggers a cascade of events involving inflammasome activation and hyperactivation of Janus kinase/signal transducer and activator of transcription signaling. Specifically, cytosolic mitochondrial DNA (mtDNA) release and caspase-1 activation are increased in FUNDC1-depleted hepatocytes. This subsequently results in the elevated release of proinflammatory cytokines, such as interleukin-1β (IL1β) and hyperproliferation of hepatocytes. Conclusion: Our results suggest that FUNDC1 suppresses HCC initiation by reducing inflammasome activation and inflammatory responses in hepatocytes, whereas up-regulation of FUNDC1 expression at the late stage of tumor development may benefit tumor growth. Our study thus describes a mechanistic link between mitophagic modulation of inflammatory response and tumorigenesis, and further implies that FUNDC1-mediated mitophagy and its related inflammatory response may represent a therapeutic target for liver cancer.
Abbreviations
-
- AIM2
-
- absent in melanoma 2
-
- ALT
-
- alanine transaminase
-
- AST
-
- aspartate transaminase
-
- ATG5
-
- autophagy related 5
-
- ATP
-
- adenosine triphosphate
-
- COX IV
-
- cytochrome c oxidase subunit IV
-
- CsA
-
- cyclosporine A
-
- DEN
-
- diethylnitrosamine
-
- EtBr
-
- ethidium bromide
-
- fl/fl
-
- FUNDC1 flox/flox mice
-
- FUNDC1
-
- FUN14 domain-containing 1
-
- FUNDC1–/–
-
- FUNDC1 whole-body knockout mice
-
- HCC
-
- hepatocellular carcinoma
-
- HCG
-
- hepatocarcinogenesis
-
- Hep FUNDC1–/–
-
- FUNDC1-depleted hepatocytes
-
- Hep WT
-
- wide-type hepatocytes
-
- Δhep
-
- FUNDC1 hepatocyte-specific knockout mice
-
- Hsp60
-
- heat shock protein 60
-
- IκB
-
- inhibitor of kappa B
-
- IHC
-
- immunohistochemistry
-
- IL1β
-
- interleukin 1β
-
- IRF3
-
- interferon regulatory factor 3
-
- JAK
-
- Janus kinase
-
- KCs
-
- Kupffer cells
-
- LC3
-
- light chain 3
-
- LF
-
- liver fibrosis
-
- LPS
-
- lipopolysaccharide
-
- mtDNA
-
- mitochondrial DNA
-
- NASH
-
- nonalcoholic steatohepatitis
-
- NF-κB
-
- nuclear factor kappa B
-
- NLRP3
-
- NLR family pyrin domain-containing 3
-
- NRF
-
- nuclear respiratory factor
-
- N.S.
-
- not significant
-
- p-
-
- phosphorylated
-
- PGC1α
-
- peroxisome proliferator-activated receptor gamma coactivator 1-alpha
-
- ROS
-
- reactive oxygen species
-
- SEM
-
- standard error of mean
-
- STAT
-
- signal transducer and activator of transcription
-
- STING
-
- stimulator of interferon genes
-
- TBK1
-
- TANK binding kinase 1
-
- TG+
-
- FUNDC1 transgenic mice
-
- Tim23
-
- translocase of inner membrane 23
-
- TG–
-
- negative FUNDC1 transgenic mice
-
- Tnfα
-
- tumor necrosis factor alpha
-
- TUNEL
-
- terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling
-
- WT
-
- wide-type mice
Hepatocellular carcinoma (HCC) accounts for most primary liver malignancies and is the most common cause of cancer-related mortality worldwide, particularly in Asia and Africa.1, 2 The pathogenesis of HCC is closely associated with chronic liver inflammation, which may result from microbial infection, toxic agents, or oxidative/metabolic stress.3 These agents or stresses converge to activate the inflammasome, a multiprotein complex which recruits adaptor protein apoptosis-associated speck-like protein containing CARD (ASC) and procaspase-1, resulting in activation of caspase-1 and cleavage of the pro-inflammatory cytokines interleukin 1β (IL1β) and IL18 in both hepatocytes and nonparenchymal cells in the liver.4, 5 These proinflammatory cytokines drive both acute and chronic liver diseases. Indeed, growing evidence shows that the inflammasome contributes to the pathogenesis of most liver diseases, including nonalcoholic steatohepatitis (NASH), steatohepatitis, hepatitis, fibrosis, and cirrhosis, and may therefore contribute to liver cancer.6, 7 In mice, liver fibrosis (LF) and HCC can be induced by a single postnatal intraperitoneal injection of the chemical carcinogen diethylnitrosamine (DEN), and DEN-induced HCC closely resembles human liver cancer.8, 9 DEN induces DNA damage and promotes cell death, which leads to an inflammatory response by resident Kupffer cells (KCs) that further hyperactivates nuclear factor kappa B (NF- B) or Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling and stimulates compensatory proliferation of hepatocytes, thus promoting tumor development.10, 11
B) or Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling and stimulates compensatory proliferation of hepatocytes, thus promoting tumor development.10, 11
Both mitochondrial dysfunction and chronic inflammation are hallmarks of cancers. Mitochondria, the bioenergetic and biosynthetic organelles, play a central role in redox homeostasis, Ca2+ signaling, innate immunity, and apoptosis.12, 13 Mitochondrial dysfunction has been linked to malignant transformation attributed to increased reactive oxygen species (ROS), altered Ca2+ signaling, dysregulated epigenetic modifications and apoptosis.14, 15 Previously, Otto Warburg proposed that mitochondrial defects explain why tumor cells preferentially undergo aerobic glycolysis.16 Mutations of genes that encode enzymes involved in mitochondrial metabolism, like succinate dehydrogenase, fumarate hydratase, isocitrate dehydrogenase 1, and isocitrate dehydrogenase 2, cause many different benign cancers.17, 18 Recent advances have suggested that when mitochondria become damaged or dysfunctional, mitochondrial ROS and mitochondrial DNA (mtDNA) are released into the cytosol to activate inflammasomes and major innate immune signaling pathways.19, 20 However, the precise mechanisms that link mitochondrial dysfunction to inflammasome activation in tumorigenesis remain poorly understood.
Mitophagy is a selective form of autophagy. It is a fundamental process for mitochondrial quality control and is required for maintenance of the mitochondrial network and reprogramming of cellular metabolism.21-23 Through selective removal of damaged or superfluous mitochondria by autophagolysosomes, mitophagy prevents accumulation of damaging mtDNA mutations and ROS. Defects in mitophagy arising from deletion, mutation, or silencing of some mitophagy adaptors, such as NIX, Parkin, and p62, lead to mitochondrial dysfunction and have been linked to inflammasome activation and cancers.24, 25 Damage to mitochondria arising from activation of the NLR family pyrin domain-containing 3 (NLRP3) inflammasome induces Parkin-dependent mitophagy that feeds back to limit inflammasome activation.25 Intriguingly, Parkin is cleaved by caspase-1 to limit mitophagy and the resultant excess inflammation.26 This two-way regulation of mitophagy and inflammation is likely to affect tumorigenesis, given the known role of inflammation in promoting cancer progression. However, this has not been directly explored. We have suggested that the mitochondrial outer-membrane protein, FUN14 domain-containing 1 (FUNDC1), is a mitophagy receptor that clears dysfunctional mitochondria in response to hypoxia and mitochondrial stresses.27, 28 We have indicated that Src kinase, the well-established oncoprotein, is able to phosphorylate FUNDC1 to prevent its binding with light chain 3 (LC3) and subsequent mitophagy.27 We were thus prompted to address the role of FUNDC1-mediated mitophagy in hepatocarcinogenesis (HCG). We found that FUNDC1 suppressed tumor initiation, and loss of FUNDC1 in hepatocytes increased susceptibility to HCG by hyperactivation of the inflammasome. In contrast, increased FUNDC1 expression during the later stage of cancer development benefits tumor growth.
Materials and Methods
Mice and Liver Tumorigenesis
Mice were maintained in the Center for Experimental Animals at the Institute of Zoology, Chinese Academy of Sciences (Beijing, China). All experimental mice were of C57BL/6 genetic background and maintained in the same conditions. FUNDC1 whole-body knockout mice were generated as described.29
Albulin-Cre mice were crossed with FUNDC1fl/fl to generate the hepatocyte-specific knockout mice (FUNDC1Δhep). To confirm FUNDC1 deletion in hepatocytes, tail genomic DNA was extracted and genotyping was done by PCR analysis using the following primers: F: 5′-GGAACAGCTCCAGATGGCAA-3′; R: 5′-AGCATGTTTAGCTGGCCCAA-3′. FUNDC1 transgenic (TG+) mice were established in Peking University (Beijing, China) and bred in the Animal Center of the Institute of Zoology, Chinese Academy of Sciences. TG+ mice and TG– mice were crossed to generate TG+ and TG– mice. Genotype of mice was confirmed by PCR using the following primers: F: 5′-GAGTGTTTGGCCACAG TTCGG-3′; R: 5′-CCAGAAGTCAGATGCTCAAGGG-3′. Mito-keima mice were crossed with FUNDC1fl/fl or FUNDC1Δhep mice to generate Mito-Keima; FUNDC1fl/fl or mito-Keima; FUNDC1Δhep mice.
For chemical induction of HCC, 15-day-old male mice and littermates were given a single intraperitoneal injection of DEN (25 mg/kg, N0258-1G; Sigma-Aldrich, St. Louis, MO) and then fed with regular chow food. Mice were sacrificed after 8 months, and livers were removed, then imaged and fixed with paraformaldehyde.
For acute liver injury, 4-week-old FUNDC1Δhepand FUNDC1fl/fl male mice were given a single intraperitoneal injection of DEN (100 mg/kg, N0258-1G; Sigma-Aldrich). These mice were sacrificed at the indicated time points (24 and 48 hours). All animals were housed in a temperature- and light-controlled animal facility, and all animal experiments were performed according to protocols approved by the Animal Care and Use Committee at the Institute of Zoology, Chinese Academy of Sciences.
Statistical Analysis
All data are presented as means ± standard error of mean (SEM). Statistical comparisons were performed with a two-tailed unpaired Student t test. The survival analysis was performed using the log-rank test. For all experiments, P values <0.05 were considered as significant (not significant [N.S.], P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001).
Detailed methodology can be found in the Supporting Materials and Methods.
Results
FUNDC1 Accumulates In Liver Tissue of Human HCC Patients
We analyzed human HCC tissue microarrays, and found that expression of FUNDC1 was significantly higher in 78% (64 of 82) of HCC samples compared to 12.5% (10 of 80) of matching peritumoral tissues (Fig. 1A). Immunoblotting analysis of HCC and peritumoral tissues from the same patients revealed that FUNDC1 expression was elevated in the tumor group compared to controls (Fig. 1B). Real-time quantitative PCR analysis showed that Fundc1 was transcriptionally up-regulated in HCC tissues (Fig. 1C). Biochemical and immunohistochemistry (IHC) analysis revealed a decrease in mitochondrial protein levels in HCC tissues compared to peritumoral tissues (Fig. 1D). mtDNA copy number, as measured by real-time PCR, was also reduced, although expression levels of the mitochondrial biogenesis factors, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1 ) and nuclear respiratory factor (NRF) 1, were not significantly changed (Fig. 1E,F). It thus appeared that level of FUNDC1 was inversely correlated with mitochondrial mass, which is likely attributed to the known role of FUNDC1 in enhancing mitochondrial clearance and/or mitochondrial turnover. Three other autophagy-related proteins, Beclin1, autophagy related 5 (ATG5), and Unc-51 like autophagy activating kinase 1, were also assayed, and they showed a similar increase in HCC tissues (Supporting Fig. S1A-D).
) and nuclear respiratory factor (NRF) 1, were not significantly changed (Fig. 1E,F). It thus appeared that level of FUNDC1 was inversely correlated with mitochondrial mass, which is likely attributed to the known role of FUNDC1 in enhancing mitochondrial clearance and/or mitochondrial turnover. Three other autophagy-related proteins, Beclin1, autophagy related 5 (ATG5), and Unc-51 like autophagy activating kinase 1, were also assayed, and they showed a similar increase in HCC tissues (Supporting Fig. S1A-D).

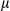 m). Heatmap of expression of FUNDC1 in human HCCs and matching peritumoral tissues (n = 82 HCCs and n = 80 peritumoral tissues). (B) Tissues were homogenized with a Dounce homogenizer and lysed in lysis buffer. Lysates from tumor and peritumoral tissues of human HCC samples (37 pairs of samples) were subjected to immunoblotting with anti-FUN DC1 antibody. FUNDC1 protein level was quantified using ImageJ software (NIH, Bethesda, MD). Results are presented as mean
m). Heatmap of expression of FUNDC1 in human HCCs and matching peritumoral tissues (n = 82 HCCs and n = 80 peritumoral tissues). (B) Tissues were homogenized with a Dounce homogenizer and lysed in lysis buffer. Lysates from tumor and peritumoral tissues of human HCC samples (37 pairs of samples) were subjected to immunoblotting with anti-FUN DC1 antibody. FUNDC1 protein level was quantified using ImageJ software (NIH, Bethesda, MD). Results are presented as mean 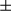 SEM; Student t test, ***P < 0.001. (C) Total RNA was extracted from tumor and peritumoral tissues (37 pairs of samples), and level of Fundc1 mRNA was determined by real-time PCR. The graph shows mean
SEM; Student t test, ***P < 0.001. (C) Total RNA was extracted from tumor and peritumoral tissues (37 pairs of samples), and level of Fundc1 mRNA was determined by real-time PCR. The graph shows mean 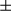 SEM; Student t test, *P < 0.05. (D) Staining of tumor and peritumor tissues from human HCC sections (10 pairs of samples) with Hsp60 and COX IV antibodies (scale bar, 100
SEM; Student t test, *P < 0.05. (D) Staining of tumor and peritumor tissues from human HCC sections (10 pairs of samples) with Hsp60 and COX IV antibodies (scale bar, 100 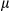 m). (E) Real-time PCR analysis of mitochondrial copy number using primers for the Cytochrome B and H19 genes (10 pairs of samples). Results are presented as mean
m). (E) Real-time PCR analysis of mitochondrial copy number using primers for the Cytochrome B and H19 genes (10 pairs of samples). Results are presented as mean 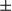 SEM; Student t test, ***P < 0.001. (F) Human liver tumor and peritumor (37 pairs of samples) extracts were homogenized. Immunoblotting analysis with antibodies against the mitochondrial proteins, Hsp60, Tim23, and COX IV, and the mitochondrial biogenesis proteins, PGC1
SEM; Student t test, ***P < 0.001. (F) Human liver tumor and peritumor (37 pairs of samples) extracts were homogenized. Immunoblotting analysis with antibodies against the mitochondrial proteins, Hsp60, Tim23, and COX IV, and the mitochondrial biogenesis proteins, PGC1 and NRF1. Levels of these proteins were quantified by ImageJ software. The graphs show mean
and NRF1. Levels of these proteins were quantified by ImageJ software. The graphs show mean 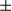 SEM; Student t test, *P < 0.05; **P < 0.01. Abbreviations: COX IV, cytochrome c oxidase subunit IV; Tim23, translocase of inner membrane 23.
SEM; Student t test, *P < 0.05; **P < 0.01. Abbreviations: COX IV, cytochrome c oxidase subunit IV; Tim23, translocase of inner membrane 23.Initiation of HCG is Promoted in FUNDC1Δhep Mice
The above results prompted us to address the role of FUNDC1 in HCG. For this purpose, liver-specific FUNDC1 knockout mice were generated by crossing FUNDC1fl/fl with Albumin-Cre transgenic mice. As revealed by western blotting and PCR analysis, FUNDC1 was specifically ablated in hepatocytes, but was present in nonparenchymal cells (Fig. 2A and Supporting Fig. S2A). We injected a single intraperitoneal dose of DEN (25 mg/kg) into 15-day-old male mice to induce HCC. This is a widely used HCC mouse model, which is considered analogous to human HCC. Most of the male mice developed typical HCC 8-10 months after DEN treatment. However, control mice, which were injected with saline, did not develop spontaneous tumors in liver even until 2 years (Supporting Fig. S2B). Analysis of serum samples collected from FUNDC1fl/fl and FUNDC1Δhep male mice 8 months after DEN treatment revealed that levels of alanine transaminase (ALT) and aspartate transaminase (AST), which are markers of hepatocellular injury or liver inflammation in most liver diseases,30 were dramatically increased in FUNDC1Δhep mice compared to controls (Fig. 2B). The number of tumors was markedly increased in FUNDC1Δhep mice compared to controls (Fig. 2C). However, maximal tumor size, which is an indicator of tumor development, was not significantly different between FUNDC1fl/fl and FUNDC1Δhep mice (Fig. 2D). These results suggest that FUNDC1 plays a critical role in the early stage of liver tumorigenesis. Furthermore, the tumor-occupied area and liver-to-body weight ratio were not significantly different in FUNDC1Δhep mice and control littermates (Fig. 2E,F).  -fetoprotein and glypican 3, markers for HCC, were positive and similar between FUNDC1-deficient tumors and tumors from FUNDC1fl/fl mice (Supporting Fig. S2C). There was no significant difference in the level of apoptotic hepatocytes, assessed by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling (TUNEL) assay, in FUNDC1fl/fl and FUNDC1Δhep 8 months after DEN treatment (Supporting Fig. S2D). Similarly, we also used a single intraperitoneal dose of DEN (25 mg/kg) to inject 15-day-old male FUNDC1 whole-body knockout mice (FUNDC1–/–) and littermates. FUNDC1–/– mice had a higher number of tumors than controls, whereas maximal tumor size was lower than in controls. Ki67 staining revealed a lower number of proliferating cells in tumor tissues of FUNDC1-null mice compared to controls, whereas the number of proliferating hepatocytes in the adjacent peritumoral tissues was higher than in control mice (Supporting Fig. S2E). Hence, ablation of FUNDC1 in hepatocytes increased the susceptibility to DEN-induced HCG compared to controls and resulted in a higher mortality rate (Supporting Fig. S2F).
-fetoprotein and glypican 3, markers for HCC, were positive and similar between FUNDC1-deficient tumors and tumors from FUNDC1fl/fl mice (Supporting Fig. S2C). There was no significant difference in the level of apoptotic hepatocytes, assessed by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling (TUNEL) assay, in FUNDC1fl/fl and FUNDC1Δhep 8 months after DEN treatment (Supporting Fig. S2D). Similarly, we also used a single intraperitoneal dose of DEN (25 mg/kg) to inject 15-day-old male FUNDC1 whole-body knockout mice (FUNDC1–/–) and littermates. FUNDC1–/– mice had a higher number of tumors than controls, whereas maximal tumor size was lower than in controls. Ki67 staining revealed a lower number of proliferating cells in tumor tissues of FUNDC1-null mice compared to controls, whereas the number of proliferating hepatocytes in the adjacent peritumoral tissues was higher than in control mice (Supporting Fig. S2E). Hence, ablation of FUNDC1 in hepatocytes increased the susceptibility to DEN-induced HCG compared to controls and resulted in a higher mortality rate (Supporting Fig. S2F).

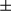 SEM; Student t test, *P < 0.05; **P < 0.01. (C) Gross morphology of livers in DEN-treated male FUNDC1fl/fl and FUNDC1Δhep mice for 8 months. Statistical analysis of tumor numbers in DEN-treated male FUNDC1fl/fl (n = 10) and FUNDC1Δhep (n = 10) mice. Results are presented as mean
SEM; Student t test, *P < 0.05; **P < 0.01. (C) Gross morphology of livers in DEN-treated male FUNDC1fl/fl and FUNDC1Δhep mice for 8 months. Statistical analysis of tumor numbers in DEN-treated male FUNDC1fl/fl (n = 10) and FUNDC1Δhep (n = 10) mice. Results are presented as mean 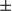 SEM; Student t test, *P < 0.05. (D) Maximum tumor size (diameters, n = 10). (E) H&E staining of liver sections from DEN-treated male FUNDC1fl/fl and FUNDC1Δhep mice. Typical liver histology (40× and 200× magnification; scale bar, 100
SEM; Student t test, *P < 0.05. (D) Maximum tumor size (diameters, n = 10). (E) H&E staining of liver sections from DEN-treated male FUNDC1fl/fl and FUNDC1Δhep mice. Typical liver histology (40× and 200× magnification; scale bar, 100 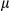 m). Black arrows indicate tumor tissue. Tumor-occupied areas in liver sections from FUNDC1fl/fl (n = 10) and FUNDC1Δhep (n = 10) mice. (F) Liver/body weight ratio 8 months after DEN injection (n = 10 for each group). Abbreviation: H&E, hematoxylin and eosin.
m). Black arrows indicate tumor tissue. Tumor-occupied areas in liver sections from FUNDC1fl/fl (n = 10) and FUNDC1Δhep (n = 10) mice. (F) Liver/body weight ratio 8 months after DEN injection (n = 10 for each group). Abbreviation: H&E, hematoxylin and eosin.FUNDC1 Deficiency Enhances Inflammatory Responses and LF
We next wished to investigate the underlying mechanism by which FUNDC1 ablation enhanced the propensity for HCG. We first carried out IHC with anti-F4/80 in HCC sections of FUNDC1fl/fl and FUNDC1Δhep mice. There was a significant increase in the number of F4/80-positive cells, which may be KCs or infiltrating macrophages in FUNDC1Δhep tumors (Fig. 3A). This observation was further confirmed by real-time PCR analysis of F4/80 mRNA level (Fig. 3B). In HCC tissues of FUNDC1Δhep mice, mRNA levels of genes encoding the inflammatory cytokines, tumor necrosis factor alpha (Tnf ), Il6, and Il1
), Il6, and Il1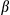 , were significantly increased. Furthermore, the absence of FUNDC1 resulted in hyperactivation of the JAK/STAT and NF-
, were significantly increased. Furthermore, the absence of FUNDC1 resulted in hyperactivation of the JAK/STAT and NF- B pathways in HCC tissues (Fig. 3C), consistent with previous findings.31, 32 Intriguingly, 1 and 2 months after DEN injection of 15-day-old male mice and before onset of cancer, the heightened JAK/STAT and NF-
B pathways in HCC tissues (Fig. 3C), consistent with previous findings.31, 32 Intriguingly, 1 and 2 months after DEN injection of 15-day-old male mice and before onset of cancer, the heightened JAK/STAT and NF- B signature was already detectable in FUNDC1Δhep liver tissues compared to controls (Fig. 3D). This suggests that the inflammatory cascade has a causal role in DEN-induced HCC. Apart from chronic liver inflammation, we also used an acute liver injury model in which 4-week-old male mice were injected intraperitoneally with DEN (100 mg/kg) for 24 or 48 hours. Deletion of FUNDC1 in hepatocytes augmented the induction of F4/80 mRNA in liver tissues, and this observation was also confirmed at the protein level by IHC detection of F4/80 (Supporting Fig. S3A). mRNA levels of Tnf
B signature was already detectable in FUNDC1Δhep liver tissues compared to controls (Fig. 3D). This suggests that the inflammatory cascade has a causal role in DEN-induced HCC. Apart from chronic liver inflammation, we also used an acute liver injury model in which 4-week-old male mice were injected intraperitoneally with DEN (100 mg/kg) for 24 or 48 hours. Deletion of FUNDC1 in hepatocytes augmented the induction of F4/80 mRNA in liver tissues, and this observation was also confirmed at the protein level by IHC detection of F4/80 (Supporting Fig. S3A). mRNA levels of Tnf , Il6, and Il1
, Il6, and Il1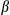 were increased in FUNDC1Δhep liver tissue in the acute model. Consistent with production of the inflammatory cytokines, Tnf
were increased in FUNDC1Δhep liver tissue in the acute model. Consistent with production of the inflammatory cytokines, Tnf and Il6, levels of phosphorylated inhibitor of kappa B (I
and Il6, levels of phosphorylated inhibitor of kappa B (I B) and STAT3 were more strongly increased in FUNDC1Δhep liver tissues than in controls (Supporting Fig. S3B). When we analyzed HCC and peritumoral tissues from DEN induced 8 months of FUNDC1–/– and WT mice, we obtained similar evidence of an enhanced inflammatory response (Supporting Fig. S3C). Taken together, these results clearly showed that FUNDC1 deficiency induced a more-severe inflammatory response in liver tissues. Moreover, FUNDC1 ablation in hepatocytes augmented fibrotic events in both tumor and peritumoral tissues from DEN-induced HCC, and this was accompanied by up-regulated expression of genes involved in fibrosis, which suggests that loss of FUNDC1 accelerated fibrosis rate (Fig. 3E). Two months after DEN administration, at an early phase in progression of HCC, degree of fibrosis and expression of fibrotic genes in FUNDC1Δhep mice were higher than in FUNDC1fl/fl mice (Fig. 3F). FUNDC1–/– mice also showed higher levels of LF (Supporting Fig. S3D). Whereas the level of NRF2 was increased in tumor tissues compared to peritumoral tissues, there was no significant difference between tumor tissues harvested from FUNDC1fl/fl (WT) and FUNDC1Δhep (FUNDC1–/–) mice (Supporting Fig. S3E,F).
B) and STAT3 were more strongly increased in FUNDC1Δhep liver tissues than in controls (Supporting Fig. S3B). When we analyzed HCC and peritumoral tissues from DEN induced 8 months of FUNDC1–/– and WT mice, we obtained similar evidence of an enhanced inflammatory response (Supporting Fig. S3C). Taken together, these results clearly showed that FUNDC1 deficiency induced a more-severe inflammatory response in liver tissues. Moreover, FUNDC1 ablation in hepatocytes augmented fibrotic events in both tumor and peritumoral tissues from DEN-induced HCC, and this was accompanied by up-regulated expression of genes involved in fibrosis, which suggests that loss of FUNDC1 accelerated fibrosis rate (Fig. 3E). Two months after DEN administration, at an early phase in progression of HCC, degree of fibrosis and expression of fibrotic genes in FUNDC1Δhep mice were higher than in FUNDC1fl/fl mice (Fig. 3F). FUNDC1–/– mice also showed higher levels of LF (Supporting Fig. S3D). Whereas the level of NRF2 was increased in tumor tissues compared to peritumoral tissues, there was no significant difference between tumor tissues harvested from FUNDC1fl/fl (WT) and FUNDC1Δhep (FUNDC1–/–) mice (Supporting Fig. S3E,F).

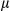 m). Statistics are shown in the bar graph. Results are presented as mean
m). Statistics are shown in the bar graph. Results are presented as mean 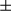 SEM; Student t test, *P < 0.05. (B) Real-time PCR of F4/80 in liver tumor tissue from FUNDC1fl/fl (n = 5) and FUNDC1Δhep (n = 5) mice. Graphs show mean
SEM; Student t test, *P < 0.05. (B) Real-time PCR of F4/80 in liver tumor tissue from FUNDC1fl/fl (n = 5) and FUNDC1Δhep (n = 5) mice. Graphs show mean 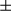 SEM; Student t test, **P < 0.01. (C) Liver tumors (n = 5 for each group) were removed for RNA extraction, and cytokine mRNA levels were determined by real-time PCR. Lysates from tumor and peritumor of DEN-treated FUNDC1fl/fl (n = 3) and FUNDC1Δhep (n = 3) mice were immunoblotted for Stat3, p-Stat3, I
SEM; Student t test, **P < 0.01. (C) Liver tumors (n = 5 for each group) were removed for RNA extraction, and cytokine mRNA levels were determined by real-time PCR. Lysates from tumor and peritumor of DEN-treated FUNDC1fl/fl (n = 3) and FUNDC1Δhep (n = 3) mice were immunoblotted for Stat3, p-Stat3, I B, and p-I
B, and p-I B. Results are presented as mean
B. Results are presented as mean 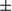 SEM; Student t test, *P < 0.05; **P < 0.01. (D) Fifteen-day-old mice were treated with DEN, then sacrificed at the indicated time points (n = 3 for each group). Lysates from livers were immunoblotted for Stat3, p-Stat3, I
SEM; Student t test, *P < 0.05; **P < 0.01. (D) Fifteen-day-old mice were treated with DEN, then sacrificed at the indicated time points (n = 3 for each group). Lysates from livers were immunoblotted for Stat3, p-Stat3, I B, and p-I
B, and p-I B. (E) Liver tissues from DEN-induced 8 months FUNDC1fl/fl (n = 6) and FUNDC1Δhep (n = 6) mice were fixed with formaldehyde. LF was analyzed by staining liver sections with Sirius Red (scale bar, 100
B. (E) Liver tissues from DEN-induced 8 months FUNDC1fl/fl (n = 6) and FUNDC1Δhep (n = 6) mice were fixed with formaldehyde. LF was analyzed by staining liver sections with Sirius Red (scale bar, 100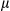 m). Positive areas were quantified with ImageJ software (NIH, Bethesda, MD). mRNA levels of fibrogenic markers (Col1a1, collagen a1(I); Col4a1, collagen a1(IV); Timp1, tissue inhibitor of metalloproteinase1; Acta2, actin a2) were determined by real-time PCR in livers. Results are presented as mean
m). Positive areas were quantified with ImageJ software (NIH, Bethesda, MD). mRNA levels of fibrogenic markers (Col1a1, collagen a1(I); Col4a1, collagen a1(IV); Timp1, tissue inhibitor of metalloproteinase1; Acta2, actin a2) were determined by real-time PCR in livers. Results are presented as mean 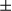 SEM; Student t test, *P < 0.05; **P < 0.01. (F) Two months of DEN-induced HCC was performed to determine LF. Sirius Red staining of liver sections from FUNDC1fl/fl (n = 3) and FUNDC1Δhep (n = 3) mice (scale bar, 100
SEM; Student t test, *P < 0.05; **P < 0.01. (F) Two months of DEN-induced HCC was performed to determine LF. Sirius Red staining of liver sections from FUNDC1fl/fl (n = 3) and FUNDC1Δhep (n = 3) mice (scale bar, 100 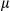 m). Sirius Red–positive areas were quantified by ImageJ software. Real-time PCR analysis of relative mRNA levels of fibrogenic markers. Results are presented as mean
m). Sirius Red–positive areas were quantified by ImageJ software. Real-time PCR analysis of relative mRNA levels of fibrogenic markers. Results are presented as mean 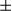 SEM; Student t test, *P < 0.05; **P < 0.01.
SEM; Student t test, *P < 0.05; **P < 0.01.Knockout of FUNDC1 Causes Accumulation of Dysfunctional Mitochondria
FUNDC1 was characterized as a receptor that mediates mitophagy, a process for eliminating dysfunctional mitochondria. In DEN-treated FUNDC1fl/fl mice, FUNDC1 protein levels were higher in tumor tissues than in matching peritumoral tissues. This is consistent with HCC patient samples. Correspondingly, mitochondrial protein levels were lower in FUNDC1fl/fl tumor tissues compared to peritumoral tissues. However, mitochondrial protein levels were higher in FUNDC1Δhep tumor tissue than in FUNDC1fl/fl tumor tissue, whereas PGC1 levels were similar in tumors from FUNDC1fl/fl and FUNDC1Δhep mice. IHC analysis of mitochondrial heat shock protein 60 (Hsp60) and real-time PCR of mtDNA further corroborated the increased mitochondrial mass in tumor tissue of FUNDC1Δhep mice (Fig. 4A and Supporting Fig. S4A). In contrast, adenosine triphosphate (ATP) levels were reduced in FUNDC1Δhep tumor tissues compared to tumor tissues of FUNDC1fl/fl mice (Fig. 4B and Supporting S4B). We selected the 1- and 2-month time points in the process of DEN-induced HCG for analysis of mitochondrial markers and functions. Mitochondrial proteins and mtDNA levels were reduced in liver tissues from FUNDC1fl/fl mice before the occurrence of HCC, and this reduction was largely blocked in liver tissues from FUNDC1Δhep mice (Fig. 4C). It is possible that DEN-induced mitochondrial defects are among the early events in HCG. To test this notion, 4-week-old FUNDC1fl/fl and FUNDC1Δhep mice were injected with a single intraperitoneal dose of DEN (100 mg/kg) for 24 or 48 hours. Immunoblotting of DEN-treated, FUNDC1-depleted liver tissues revealed higher levels of mitochondrial proteins, indicative of greater accumulation of mitochondria, compared to controls (Supporting Fig. S4C). This result was also confirmed by real-time PCR to detect mtDNA and by IHC to detect Hsp60 (Supporting Fig. S4D). Mitochondrial functions, as measured by ATP production and oxygen consumption rate, were assessed in livers of FUNDC1Δhep and FUNDC1fl/fl mice. Indeed, there was a 20% decrease in levels of ATP and respiration in FUNDC1Δhep liver tissues following DEN treatment (Supporting Fig. S4E). Surprisingly, loss of FUNDC1 reduced the production of ATP, even though mitochondrial protein levels were much higher in liver tissues. We further investigated whether the mitochondrial defects are attributed to impair FUNDC1-mediated mitophagy in hepatocytes. Mitochondrial marker proteins were decreased in isolated hepatocytes from FUNDC1fl/fl mice treated with acute DEN (100 mg/kg), whereas the protein degradation was inhibited to a certain extent in isolated hepatocytes from FUNDC1Δhep liver tissue. However, there was no difference in levels of mitochondrial proteins in isolated resident KCs from acute DEN-treated WT and FUNDC1–/– mice (Supporting Fig. S4F). To further demonstrate that FUNDC1 mediates mitophagy in hepatocytes in response to DEN treatment, we bred mito-keima mice with FUNDC1fl/fl and FUNDC1Δhep mice and isolated and cultured hepatocytes from these mito-keima–labeled primary hepatocytes in the collagen-coated, glass-bottom cell-culture dish. DEN treatment significantly increased the appearance of red puncta, the marker of the mitophagy,33 in Hep WT (wild-type hepatocytes), but not in FUNDC1-depleted hepatocytes (Fig. 4D). We further compared levels of LC3 and p62, which are biochemical hallmarks of autophagy, between mitochondria isolated from hepatocytes from acute DEN-treated FUNDC1fl/fl and FUNDC1Δhep mice. We found that there were higher levels of LC3 and lower levels of p62 in FUNDC1fl/fl hepatocellular mitochondria than in FUNDC1Δhep hepatocellular mitochondria (Fig. 4E). Electron microscopy analysis confirmed that acute DEN treatment induced the appearance of mitophagosomes, and FUNDC1 ablation blocked mitophagosome formation (Fig. 4F).
levels were similar in tumors from FUNDC1fl/fl and FUNDC1Δhep mice. IHC analysis of mitochondrial heat shock protein 60 (Hsp60) and real-time PCR of mtDNA further corroborated the increased mitochondrial mass in tumor tissue of FUNDC1Δhep mice (Fig. 4A and Supporting Fig. S4A). In contrast, adenosine triphosphate (ATP) levels were reduced in FUNDC1Δhep tumor tissues compared to tumor tissues of FUNDC1fl/fl mice (Fig. 4B and Supporting S4B). We selected the 1- and 2-month time points in the process of DEN-induced HCG for analysis of mitochondrial markers and functions. Mitochondrial proteins and mtDNA levels were reduced in liver tissues from FUNDC1fl/fl mice before the occurrence of HCC, and this reduction was largely blocked in liver tissues from FUNDC1Δhep mice (Fig. 4C). It is possible that DEN-induced mitochondrial defects are among the early events in HCG. To test this notion, 4-week-old FUNDC1fl/fl and FUNDC1Δhep mice were injected with a single intraperitoneal dose of DEN (100 mg/kg) for 24 or 48 hours. Immunoblotting of DEN-treated, FUNDC1-depleted liver tissues revealed higher levels of mitochondrial proteins, indicative of greater accumulation of mitochondria, compared to controls (Supporting Fig. S4C). This result was also confirmed by real-time PCR to detect mtDNA and by IHC to detect Hsp60 (Supporting Fig. S4D). Mitochondrial functions, as measured by ATP production and oxygen consumption rate, were assessed in livers of FUNDC1Δhep and FUNDC1fl/fl mice. Indeed, there was a 20% decrease in levels of ATP and respiration in FUNDC1Δhep liver tissues following DEN treatment (Supporting Fig. S4E). Surprisingly, loss of FUNDC1 reduced the production of ATP, even though mitochondrial protein levels were much higher in liver tissues. We further investigated whether the mitochondrial defects are attributed to impair FUNDC1-mediated mitophagy in hepatocytes. Mitochondrial marker proteins were decreased in isolated hepatocytes from FUNDC1fl/fl mice treated with acute DEN (100 mg/kg), whereas the protein degradation was inhibited to a certain extent in isolated hepatocytes from FUNDC1Δhep liver tissue. However, there was no difference in levels of mitochondrial proteins in isolated resident KCs from acute DEN-treated WT and FUNDC1–/– mice (Supporting Fig. S4F). To further demonstrate that FUNDC1 mediates mitophagy in hepatocytes in response to DEN treatment, we bred mito-keima mice with FUNDC1fl/fl and FUNDC1Δhep mice and isolated and cultured hepatocytes from these mito-keima–labeled primary hepatocytes in the collagen-coated, glass-bottom cell-culture dish. DEN treatment significantly increased the appearance of red puncta, the marker of the mitophagy,33 in Hep WT (wild-type hepatocytes), but not in FUNDC1-depleted hepatocytes (Fig. 4D). We further compared levels of LC3 and p62, which are biochemical hallmarks of autophagy, between mitochondria isolated from hepatocytes from acute DEN-treated FUNDC1fl/fl and FUNDC1Δhep mice. We found that there were higher levels of LC3 and lower levels of p62 in FUNDC1fl/fl hepatocellular mitochondria than in FUNDC1Δhep hepatocellular mitochondria (Fig. 4E). Electron microscopy analysis confirmed that acute DEN treatment induced the appearance of mitophagosomes, and FUNDC1 ablation blocked mitophagosome formation (Fig. 4F).

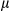 m). mtDNA copy number was determined by real-time PCR. PGC1
m). mtDNA copy number was determined by real-time PCR. PGC1 , Hsp60, VDAC1, Tim23, COX IV, and FUNDC1 proteins were analyzed by western blotting. Results are presented as mean
, Hsp60, VDAC1, Tim23, COX IV, and FUNDC1 proteins were analyzed by western blotting. Results are presented as mean 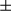 SEM; Student t test, ***P < 0.001. (B) Representative ATP levels are shown in tumor and peritumoral tissues from FUNDC1fl/fl and FUNDC1Δhep mice (n = 6 for each group). Results are presented as mean
SEM; Student t test, ***P < 0.001. (B) Representative ATP levels are shown in tumor and peritumoral tissues from FUNDC1fl/fl and FUNDC1Δhep mice (n = 6 for each group). Results are presented as mean 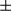 SEM; Student t test, *P < 0.05; **P < 0.01. (C) Fifteen-day-old FUNDC1fl/fl and FUNDC1Δhep mice were treated with DEN (25 mg/kg), and then sacrificed at 1 and 2 months. mtDNA copy number were quantified by real-time PCR in livers. Mitochondrial proteins were assessed by immunoblotting analysis in livers. Results are presented as mean
SEM; Student t test, *P < 0.05; **P < 0.01. (C) Fifteen-day-old FUNDC1fl/fl and FUNDC1Δhep mice were treated with DEN (25 mg/kg), and then sacrificed at 1 and 2 months. mtDNA copy number were quantified by real-time PCR in livers. Mitochondrial proteins were assessed by immunoblotting analysis in livers. Results are presented as mean 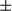 SEM; Student t test, **P < 0.01; ***P < 0.001. (D) Hepatocytes were isolated from mito-keima; FUNDC1fl/fl and mito-keima; FUNDC1Δhep mice. After DEN (500
SEM; Student t test, **P < 0.01; ***P < 0.001. (D) Hepatocytes were isolated from mito-keima; FUNDC1fl/fl and mito-keima; FUNDC1Δhep mice. After DEN (500 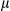 g/mL) treatment for 24 hours, live hepatocytes were observed. Green structures are consistent with mitochondria, and individual red puncta is wholly contained within the lysosome. Results are presented as mean
g/mL) treatment for 24 hours, live hepatocytes were observed. Green structures are consistent with mitochondria, and individual red puncta is wholly contained within the lysosome. Results are presented as mean 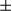 SEM; Student t test, **P < 0.01 (n = 3). (E) Hepatocytes were isolated from acute DEN-treated (100 mg/kg) FUNDC1fl/fl and FUNDC1Δhep mice (n = 3 for each group) for 24 and 48 hours, and mitochondria were isolated from hepatocytes. Western blotting analysis of p62 and LC3 levels in hepatocytes and mitochondria. (F) Electron micrographs of mitochondria and autophagosomes in liver tissues 24 hours after DEN treatment. The number of mitochondria incorporated into autophagosome was quantified from more than three different liver sections of each group. Results are presented as mean
SEM; Student t test, **P < 0.01 (n = 3). (E) Hepatocytes were isolated from acute DEN-treated (100 mg/kg) FUNDC1fl/fl and FUNDC1Δhep mice (n = 3 for each group) for 24 and 48 hours, and mitochondria were isolated from hepatocytes. Western blotting analysis of p62 and LC3 levels in hepatocytes and mitochondria. (F) Electron micrographs of mitochondria and autophagosomes in liver tissues 24 hours after DEN treatment. The number of mitochondria incorporated into autophagosome was quantified from more than three different liver sections of each group. Results are presented as mean 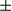 SEM; Student t test, *P < 0.05. Abbreviations: Cont, control; COX IV, cytochrome c oxidase subunit IV; VDAC1, voltage-dependent anion-selective channel 1; Tim23, translocase of inner membrane 23.
SEM; Student t test, *P < 0.05. Abbreviations: Cont, control; COX IV, cytochrome c oxidase subunit IV; VDAC1, voltage-dependent anion-selective channel 1; Tim23, translocase of inner membrane 23.Loss of FUNDC1 Aggravates Activation of Caspase-1
Our findings indicate that DEN elicits both chronic inflammatory responses and acute mitochondrial stress responses, including loss of mitochondrial functions and enhanced mitophagy. We were interested to understand how the acute mitochondrial stresses lead to chronic inflammatory response. Because mitophagy is critical for removal of dysfunctional or damaged mitochondria, which are implicated in the activation of inflammatory responses,34 we examined the effect of FUNDC1 deficiency on activation of caspase-1 in hepatocytes upon DEN treatment. FUNDC1Δhep and FUNDC1fl/fl mice were injected with DEN (100 mg/kg), which were sacrificed 24 or 48 hours later. FUNDC1-deficient hepatocytes had higher levels of the cleaved form of caspase-1 than Hep WT in response to DEN treatment in vivo. Additionally, mRNA levels of Nlrp3 and Il1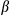 were increased in FUNDC1-depleted hepatocytes, and treatment of FUNDC1Δhep mice with DEN also enhanced the level of IL1
were increased in FUNDC1-depleted hepatocytes, and treatment of FUNDC1Δhep mice with DEN also enhanced the level of IL1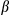 in serum samples collected at 24 and 48 hours postinjection (Supporting Fig. S5A). To investigate the inflammatory phenotype in the early stages of HCG, we analyzed mice 1 month after DEN-treated (25 mg/kg) 15-day-old male mice. As expected, hepatocytes isolated from liver tissues of FUNDC1Δhep mice 1 month after DEN injection showed evidence of mitochondrial accumulation (Supporting Fig. S5B). mRNA levels of Nlrp3 and Il1
in serum samples collected at 24 and 48 hours postinjection (Supporting Fig. S5A). To investigate the inflammatory phenotype in the early stages of HCG, we analyzed mice 1 month after DEN-treated (25 mg/kg) 15-day-old male mice. As expected, hepatocytes isolated from liver tissues of FUNDC1Δhep mice 1 month after DEN injection showed evidence of mitochondrial accumulation (Supporting Fig. S5B). mRNA levels of Nlrp3 and Il1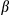 in DEN-treated FUNDC1Δhep liver tissues were also increased, which is indicative of inflammasome activation (Fig. 5A). We further assessed the cleaved form of caspase-1 in 1-month DEN treated FUNDC1Δhep and FUNDC1fl/fl mice. Interestingly, levels of cleavage of caspase-1 were higher in FUNDC1-deficient hepatocytes; however, caspase-1 activation was not different in both WT and FUNDC1-depleted KCs from WT and FUNDC1–/– animals (Fig. 5B). This result is also confirmed by using the FLICA caspase-1 assay25 to detect active caspase-1 in isolated hepatocytes and KCs from DEN-treated 1-month WT and FUNDC1–/– mice (Fig. 5C). DEN specifically activates the inflammasome, as measured by caspase-1 cleavage; similar results were confirmed in primary hepatocytes isolated from mice 2 and 5 months after DEN (25 mg/kg) treatment (Supporting Fig. S5C). However, the stimulator of interferon genes (STING) pathway was not strongly affected because the phosphorylated TANK binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3), the downstream of STING, were not induced in either hepatocytes or KCs isolated from mice 1 month after DEN treatment (Supporting Fig. S5D). DEN treatment enhanced caspase-1/NLRP3 or caspase-1/absent in melanoma 2 (AIM2) complex formation in vivo, which was further augmented in FUNDC1-depleted hepatocytes (Fig. 5D). This is further substantiated by the more-pronounced cleavage of caspase-1 and matured IL1
in DEN-treated FUNDC1Δhep liver tissues were also increased, which is indicative of inflammasome activation (Fig. 5A). We further assessed the cleaved form of caspase-1 in 1-month DEN treated FUNDC1Δhep and FUNDC1fl/fl mice. Interestingly, levels of cleavage of caspase-1 were higher in FUNDC1-deficient hepatocytes; however, caspase-1 activation was not different in both WT and FUNDC1-depleted KCs from WT and FUNDC1–/– animals (Fig. 5B). This result is also confirmed by using the FLICA caspase-1 assay25 to detect active caspase-1 in isolated hepatocytes and KCs from DEN-treated 1-month WT and FUNDC1–/– mice (Fig. 5C). DEN specifically activates the inflammasome, as measured by caspase-1 cleavage; similar results were confirmed in primary hepatocytes isolated from mice 2 and 5 months after DEN (25 mg/kg) treatment (Supporting Fig. S5C). However, the stimulator of interferon genes (STING) pathway was not strongly affected because the phosphorylated TANK binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3), the downstream of STING, were not induced in either hepatocytes or KCs isolated from mice 1 month after DEN treatment (Supporting Fig. S5D). DEN treatment enhanced caspase-1/NLRP3 or caspase-1/absent in melanoma 2 (AIM2) complex formation in vivo, which was further augmented in FUNDC1-depleted hepatocytes (Fig. 5D). This is further substantiated by the more-pronounced cleavage of caspase-1 and matured IL1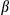 in cultured primary hepatocytes isolated from FUNDC1Δhep treated with lipopolysaccharide (LPS) and DEN (100 or 500 µg/mL) compared to FUNDC1fl/fl mice (Fig. 5E,F). In summary, FUNDC1 ablation promoted accumulation of dysfunctional mitochondria, and mitophagy reduced, but did not completely prevent, inflammasome activation.
in cultured primary hepatocytes isolated from FUNDC1Δhep treated with lipopolysaccharide (LPS) and DEN (100 or 500 µg/mL) compared to FUNDC1fl/fl mice (Fig. 5E,F). In summary, FUNDC1 ablation promoted accumulation of dysfunctional mitochondria, and mitophagy reduced, but did not completely prevent, inflammasome activation.

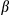 , was assessed by real-time PCR of total mRNA isolated from hepatocytes of FUNDC1fl/fl and FUNDC1Δhep mice (n = 3 for each group) 1 month after DEN (25 mg/kg) treatment. Results are presented as mean
, was assessed by real-time PCR of total mRNA isolated from hepatocytes of FUNDC1fl/fl and FUNDC1Δhep mice (n = 3 for each group) 1 month after DEN (25 mg/kg) treatment. Results are presented as mean 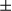 SEM; Student t test, *P < 0.05; **P < 0.01. (B) Western blotting analysis of procaspase-1 and p20-caspase-1 (cleaved caspase-1) in isolated hepatocytes (FUNDC1fl/fl and FUNDC1Δhep mice) and KCs (WT and FUNDC1–/– mice) 1 month after DEN (25 mg/kg) treatment (n = 3 for each group). (C) Caspase-1 activity in hepatocytes and KCs isolated from 1-month DEN-treated WT and FUNDC1–/– mice (n = 3 for each goup). Results are presented as mean
SEM; Student t test, *P < 0.05; **P < 0.01. (B) Western blotting analysis of procaspase-1 and p20-caspase-1 (cleaved caspase-1) in isolated hepatocytes (FUNDC1fl/fl and FUNDC1Δhep mice) and KCs (WT and FUNDC1–/– mice) 1 month after DEN (25 mg/kg) treatment (n = 3 for each group). (C) Caspase-1 activity in hepatocytes and KCs isolated from 1-month DEN-treated WT and FUNDC1–/– mice (n = 3 for each goup). Results are presented as mean 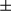 SEM; Student t test, **P < 0.01. (D) Caspase-1 interacts with NLRP3 and AIM2 in WT and FUNDC1-depleted hepatocytes isolated from the acute liver injury model (n = 3 for each group). (E) Western blotting analysis of p20-caspase-1 in LPS-primed hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice (n = 5 for each group) stimulated with DEN (100
SEM; Student t test, **P < 0.01. (D) Caspase-1 interacts with NLRP3 and AIM2 in WT and FUNDC1-depleted hepatocytes isolated from the acute liver injury model (n = 3 for each group). (E) Western blotting analysis of p20-caspase-1 in LPS-primed hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice (n = 5 for each group) stimulated with DEN (100 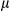 g/mL, 500
g/mL, 500 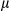 g/mL) for 24 hours. (F) Secretion of IL
g/mL) for 24 hours. (F) Secretion of IL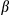 by the above cells was measured by enzyme-linked immunosorbent assay. Results are presented as mean
by the above cells was measured by enzyme-linked immunosorbent assay. Results are presented as mean  SEM; Student t test, *P < 0.05. Abbreviations: Cont, control; IgG, immunoglobulin G; IP, immunoprecipitation; KO, knockout.
SEM; Student t test, *P < 0.05. Abbreviations: Cont, control; IgG, immunoglobulin G; IP, immunoprecipitation; KO, knockout.Previous findings have reported that IL1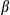 released from activated hepatocytes or KCs is able to promote hepatocellular proliferation.35, 36 We found that mRNAs of Tnf
released from activated hepatocytes or KCs is able to promote hepatocellular proliferation.35, 36 We found that mRNAs of Tnf and Il6 were increased in KCs following incubation with the conditioned medium from ATP-treated, LPS-primed hepatocytes. As expected, such induction is higher when KCs are incubated with medium from FUNDC1-depleted hepatocytes. On the other hand, when the conditioned medium from aforementional KCs was used to treat isolated hepatocytes, levels of phosphorylated (p)-Stat3 and p-I
and Il6 were increased in KCs following incubation with the conditioned medium from ATP-treated, LPS-primed hepatocytes. As expected, such induction is higher when KCs are incubated with medium from FUNDC1-depleted hepatocytes. On the other hand, when the conditioned medium from aforementional KCs was used to treat isolated hepatocytes, levels of phosphorylated (p)-Stat3 and p-I B were significantly enhanced, and such activation is more pronounced in FUNDC1-depleted hepatocytes (Supporting Fig. S5E). Importantly, addition of IL1
B were significantly enhanced, and such activation is more pronounced in FUNDC1-depleted hepatocytes (Supporting Fig. S5E). Importantly, addition of IL1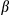 antibody into the conditioned medium blocked induction of mRNAs of Tnf
antibody into the conditioned medium blocked induction of mRNAs of Tnf and Il6 in KCs and prevented hepatocellular proliferation, as revealed by the 5-bromo-2-deoxyuridine incorporation analysis (Supporting Fig. S5F). It would be logical to assume that FUNDC1 depletion in hepatocytes results in greater production of mature IL1
and Il6 in KCs and prevented hepatocellular proliferation, as revealed by the 5-bromo-2-deoxyuridine incorporation analysis (Supporting Fig. S5F). It would be logical to assume that FUNDC1 depletion in hepatocytes results in greater production of mature IL1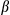 . It can stimulate KCs and produces more IL6 and TNF
. It can stimulate KCs and produces more IL6 and TNF , which, in turn, up-regulates the signaling pathways in hepatocytes.
, which, in turn, up-regulates the signaling pathways in hepatocytes.
Cytosolic Mitochondrial DNA is Required For Caspase-1 Activation and IL1β Release
It has been reported that release of mtDNA and mitochondrial ROS activate the inflammasome, which, in turn, promotes the maturation of IL1β.19, 20 Therefore, we further investigated the mechanistic link between cytosolic mtDNA and inflammasome activation in hepatocytes stimulated with LPS and DEN. Indeed, treatment with DEN led to an increase in cytosolic mitochondrial DNA in LPS-primed, FUNDC1-depleted primary hepatocytes (Fig. 6A). Level of secretory mature IL1β was also increased in FUNDC1-depleted primary hepatocytes, and this increase was dramatically reduced by cyclosporine A (CsA; Fig. 6B). In LPS-primed hepatocytes, ATP-driven activation of caspase-1 is a stronger inflammasome activator than DEN treatment. Therefore, we treated primary hepatocytes from FUNDC1Δhep and FUNDC1fl/fl mice with LPS and ATP and assayed the cytosolic mtDNA. Level of cytosolic mtDNA was higher in ATP-treated, LPS-primed FUNDC1Δhep primary hepatocytes than in control hepatocytes. This result was also confirmed by confocal microscopy, using antibodies detecting DNA and the mitochondrial matrix protein, Hsp60 (Fig. 6C). However, it is noteworthy that baseline expression of FUNDC1 was lower in KCs compared to that of hepatocytes. Moreover, treatment of WT and FUNDC1-depleted KCs with LPS and ATP or poly(dA:dT) led to release of statistically indistinguishable levels of mature IL1 β between both (Supporting Fig. S6A,B). Release of mtDNA is likely attributed to mitochondrial damages, given that inhibition of the mitochondrial permeability transition pore (MPTP) by CsA prevented inflammasome activation and reduced cleavage of IL1β in LPS-primed WT and FUNDC1-depleted hepatocytes stimulated with ATP or poly(dA:dT) (Fig. 6D). This finding was further confirmed with ethidium bromide (EtBr) treatment, which diminishes the level of mtDNA in hepatocytes.25 Incubation with EtBr inhibited inflammasome activation in both WT and FUNDC1-depleted hepatocytes (Fig. 6E and Supporting Fig. S6C). Transfection of an FUNDC1-expressing plasmid into FUNDC1-depleted hepatocytes prevented inflammasome activation, as indicated by reduced IL1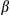 cleavage, whereas expression of a mitophagy-defective FUNDC1 ΔLIR mutant, which lacks the LC3-interacting region, only partially rescued activation upon treatment of LPS-primed, FUNDC1-depleted hepatocytes with ATP or poly(dA:dT) (Fig. 6F). These results showed that ablation of FUNDC1 promoted mitochondrial DNA release and activation of the inflammasome in a manner that depended on FUNDC1-mediated mitophagy.
cleavage, whereas expression of a mitophagy-defective FUNDC1 ΔLIR mutant, which lacks the LC3-interacting region, only partially rescued activation upon treatment of LPS-primed, FUNDC1-depleted hepatocytes with ATP or poly(dA:dT) (Fig. 6F). These results showed that ablation of FUNDC1 promoted mitochondrial DNA release and activation of the inflammasome in a manner that depended on FUNDC1-mediated mitophagy.

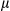 g/mL, 500
g/mL, 500 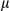 g/mL). (B) LPS-primed hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice were incubated with CsA (10
g/mL). (B) LPS-primed hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice were incubated with CsA (10  M), then stimulated with ATP (5 mM) and DEN (500
M), then stimulated with ATP (5 mM) and DEN (500 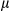 g/ml). IL1
g/ml). IL1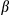 secretion was measured by enzyme-linked immunosorbent assay (ELISA). (C) LPS-primed hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice were incubated with ATP. (I) Quantitative PCR analysis of cytosolic mtDNA in hepatocytes. (II) Confocal microscopy of LPS-primed hepatocytes treated for 3 hours with ATP, then immunostained for Hsp60 (red) and DNA (green). Yellow dots indicate colocalization of red (mitochondria) and green (DNA) signals. (D) Cultured primary hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice were incubated with CsA (10
secretion was measured by enzyme-linked immunosorbent assay (ELISA). (C) LPS-primed hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice were incubated with ATP. (I) Quantitative PCR analysis of cytosolic mtDNA in hepatocytes. (II) Confocal microscopy of LPS-primed hepatocytes treated for 3 hours with ATP, then immunostained for Hsp60 (red) and DNA (green). Yellow dots indicate colocalization of red (mitochondria) and green (DNA) signals. (D) Cultured primary hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice were incubated with CsA (10 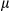 M), then treated with LPS and stimulated with ATP or transfected with poly(dA:dT) (1.5
M), then treated with LPS and stimulated with ATP or transfected with poly(dA:dT) (1.5 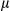 g/ml). The top panel shows ELISA analysis of IL1
g/ml). The top panel shows ELISA analysis of IL1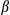 secretion. The bottom panel shows immunoblotting analysis of IL1
secretion. The bottom panel shows immunoblotting analysis of IL1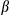 in the supernatants. (E) Cultured primary hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice were exposed to EtBr (450 ng/mL, 72 hours), then treated with LPS and stimulated with ATP or transfected with poly(dA:dT). The top panel shows ELISA analysis of IL1
in the supernatants. (E) Cultured primary hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice were exposed to EtBr (450 ng/mL, 72 hours), then treated with LPS and stimulated with ATP or transfected with poly(dA:dT). The top panel shows ELISA analysis of IL1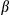 secretion. The bottom panel shows immunoblotting analysis of IL1
secretion. The bottom panel shows immunoblotting analysis of IL1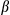 in supernatants. (F) Cultured primary hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice were transfected with FUNDC1 and FUNDC1
in supernatants. (F) Cultured primary hepatocytes isolated from FUNDC1fl/fl and FUNDC1Δhep mice were transfected with FUNDC1 and FUNDC1  LIR plasmids, then treated with LPS and stimulated with ATP. The top panel shows ELISA measurement of IL1
LIR plasmids, then treated with LPS and stimulated with ATP. The top panel shows ELISA measurement of IL1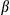 secretion. The bottom panel shows immunoblotting analysis of IL1
secretion. The bottom panel shows immunoblotting analysis of IL1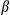 in supernatants. Results in (A), (B), (C), (D), (E), and (F) are presented as mean
in supernatants. Results in (A), (B), (C), (D), (E), and (F) are presented as mean  SEM; Student t test, *P < 0.05; **P < 0.01; ***P < 0.001.
SEM; Student t test, *P < 0.05; **P < 0.01; ***P < 0.001.HCG is Decreased in FUNDC1 Transgenic Mice
To further explore the role of FUNDC1 in HCC, we constructed FUNDC1 transgenic mice, which overexpressed FUNDC1 in hepatocytes (Supporting Fig. S6D). We administered a single injection of DEN to 15-day-old male mice to induce HCC. Strikingly, the number of detectable tumor nodes was lower in FUNDC1 transgenic mice than controls (Fig. 7A). Ratio of liver weight and body weight was also decreased in FUNDC1 transgenic mice (Fig. 7B). Surprisingly, mitochondrial proteins and mtDNA were not obviously decreased in HCC tissues from FUNDC1 transgenic mice. However, the mitochondrial biogenesis protein, PGC1 , was maintained at a high level in FUNDC1 transgenic HCC tissues (Fig. 7C). As expected, the number of F4/80-positive cells was decreased in livers of FUNDC1 transgenic mice, and mRNA levels of Tnf
, was maintained at a high level in FUNDC1 transgenic HCC tissues (Fig. 7C). As expected, the number of F4/80-positive cells was decreased in livers of FUNDC1 transgenic mice, and mRNA levels of Tnf and Il6 were also decreased in FUNDC1 transgenic HCCs compared to controls. Lower expression of p-Stat3 and a higher level of p-I
and Il6 were also decreased in FUNDC1 transgenic HCCs compared to controls. Lower expression of p-Stat3 and a higher level of p-I B also confirmed that FUNDC1 overexpression in transgenic mice reduced the downstream signaling pathways, which was opposite to the effect observed in control mice (Fig. 7D). We also cultured isolated primary hepatocytes from FUNDC1 transgenic mice and littermate mice, and treated them with LPS, ATP, and poly(dA:dT) in vitro. FUNDC1 overexpression reduced the level of cleaved IL1β and inhibited inflammasome activation (Fig. 7E).
B also confirmed that FUNDC1 overexpression in transgenic mice reduced the downstream signaling pathways, which was opposite to the effect observed in control mice (Fig. 7D). We also cultured isolated primary hepatocytes from FUNDC1 transgenic mice and littermate mice, and treated them with LPS, ATP, and poly(dA:dT) in vitro. FUNDC1 overexpression reduced the level of cleaved IL1β and inhibited inflammasome activation (Fig. 7E).

 SEM; Student t test, *P < 0.05. H&E staining of WT and FUNDC1 transgenic liver sections (scale bar, 100
SEM; Student t test, *P < 0.05. H&E staining of WT and FUNDC1 transgenic liver sections (scale bar, 100 m). Black arrows indicate tumor tissue. (B) Liver/body weight ratios were determined for the two groups of mice (n = 8 for each group). Results are presented as mean
m). Black arrows indicate tumor tissue. (B) Liver/body weight ratios were determined for the two groups of mice (n = 8 for each group). Results are presented as mean  SEM; Student t test, **P < 0.01. (C) Tumor and peritumoral tissues were homogenized and extracted proteins and total genomes (n = 3 for each group). (I) mtDNA copy numbers were measured by real-time PCR. Results are shown as mean
SEM; Student t test, **P < 0.01. (C) Tumor and peritumoral tissues were homogenized and extracted proteins and total genomes (n = 3 for each group). (I) mtDNA copy numbers were measured by real-time PCR. Results are shown as mean  SEM; Student t test, *P < 0.05. (II) Expression of PGC1
SEM; Student t test, *P < 0.05. (II) Expression of PGC1 , Hsp60, VDAC1, Tim23, and COX IV proteins in tumor and peritumor of WT and FUNDC1 transgenic mice. (D) Inflammatory response was determined in tumor and peritumoral tissues by IHC, real-time PCR, and western blotting (n = 3 for each goup). (I) Representative anti-F4/80 staining of liver sections of WT and FUNDC1 transgenic HCCs showing F4/80 expression in KCs/liver macrophages (scale bar, 100
, Hsp60, VDAC1, Tim23, and COX IV proteins in tumor and peritumor of WT and FUNDC1 transgenic mice. (D) Inflammatory response was determined in tumor and peritumoral tissues by IHC, real-time PCR, and western blotting (n = 3 for each goup). (I) Representative anti-F4/80 staining of liver sections of WT and FUNDC1 transgenic HCCs showing F4/80 expression in KCs/liver macrophages (scale bar, 100  m). (II) F4/80 mRNA levels in tumor tissues were quantified by real-time PCR. Results are shown as mean
m). (II) F4/80 mRNA levels in tumor tissues were quantified by real-time PCR. Results are shown as mean 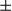 SEM; Student t test, *P < 0.05. (III) Expression levels of the indicated genes were determined by real-time PCR. Results are presented as mean
SEM; Student t test, *P < 0.05. (III) Expression levels of the indicated genes were determined by real-time PCR. Results are presented as mean 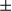 SEM; Student t test, *P < 0.05; **P < 0.01. (IV) Immunoblotting was performed on liver lysates of tumor and peritumoral tissues. p-Stat3 and p-I
SEM; Student t test, *P < 0.05; **P < 0.01. (IV) Immunoblotting was performed on liver lysates of tumor and peritumoral tissues. p-Stat3 and p-I B were assessed. (E) LPS-primed WT and FUNDC1 transgenic hepatocytes were treated with ATP or transfected with poly(dA:dT). IL1β secretion was measured by enzyme-linked immunosorbent assay. Results are shown as mean
B were assessed. (E) LPS-primed WT and FUNDC1 transgenic hepatocytes were treated with ATP or transfected with poly(dA:dT). IL1β secretion was measured by enzyme-linked immunosorbent assay. Results are shown as mean 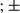 SEM; Student t test, *P < 0.05; **P < 0.01. (F) A model explaining our results. The inflammasome is activated by mtDNA and mtROS released by damaged mitochondria under mitochondrial stress (induced by DEN) in hepatocytes. This process is partially, but not completely, prevented by FUNDC1-mediated mitophagy. In FUNDC1-depleted hepatocytes, accumulated mitochondria release more mtDNA and mtROS, which hyperactivates the inflammasome and leads to proliferation of mutant hepatocytes. Abbreviations: Cont, control; COX IV, cytochrome c oxidase subunit IV; VDAC1, voltage-dependent anion-selective channel 1; Tim23, translocase of inner membrane 23.
SEM; Student t test, *P < 0.05; **P < 0.01. (F) A model explaining our results. The inflammasome is activated by mtDNA and mtROS released by damaged mitochondria under mitochondrial stress (induced by DEN) in hepatocytes. This process is partially, but not completely, prevented by FUNDC1-mediated mitophagy. In FUNDC1-depleted hepatocytes, accumulated mitochondria release more mtDNA and mtROS, which hyperactivates the inflammasome and leads to proliferation of mutant hepatocytes. Abbreviations: Cont, control; COX IV, cytochrome c oxidase subunit IV; VDAC1, voltage-dependent anion-selective channel 1; Tim23, translocase of inner membrane 23.Discussion
In the present study, we have uncovered the dual role of FUNDC1, a well-characterized mitophagy receptor, in the initiation and progression of HCC. It has a suppressive role in the initiation of HCG, because deletion of FUNDC1 resulted in increased development of HCC and higher mortality, whereas overexpression of FUNDC1 had antitumor effects. The increased tumor numbers, albeit unaffected tumor size, in FUNDC1Δhep mice further support the view that FUNDC1 may suppress initiation of HCC. Mitophagy is considered a physiological fail-safe mechanism, which maintains an intact and functionally dependable mitochondrial network. Damaged mitochondria are powder kegs of mitochondrial integrity, mutations of mitochondrial genomes, dysfunctional mitochondrial metabolisms, and heightened ROS production, which were associated with liver tumorigenesis. Knockout of essential mitophagy genes like Parkin increased susceptibility to liver tumors, and BCL2 interacting protein 3 promoted mammary tumor progression.37, 38 We identified that FUNDC1 expression is higher in human HCC tissues than peritumoral tissues in 78% of samples investigated, which is consistent with a recent report showing that FUNDC1 is highly expressed in cervical cancers.39 These data suggest that up-regulation of FUNDC1 expression is required for and benefits the late stage of tumor development. It is thus proposed that mitophagy, a selective form of macroautophagy, has dual roles in cancer.40 Our findings are consistent with the established dual role of general autophagy in cancer. Autophagy is an evolutionarily conserved pathway for clearance of damaged or harmful proteins and organelles. Autophagy defects can cause genetic instability, increased expression of p62, and accumulation of abnormal mitochondria. For instance, allelic loss of the essential autophagy gene, Beclin 1 (ATG6), augmented human breast, ovarian, and spontaneous liver tumors.41 Mice with deletion of ATG5 and ATG7 developed liver adenomas attributed to mitochondrial damage and genomic damage responses.42 During the late stage of tumor development, autophagy also satisfies the high metabolic demands of proliferating tumor cells.43
We have demonstrated that FUNDC1 reduces inflammasome activation through its regulatory effect on mitophagy. FUNDC1-ablated hepatocytes showed mitochondrial accumulation, reduced ATP production, and lower oxygen consumption without obviously altered mitochondrial biogenesis. After acute DEN injection, liver tissue from FUNDC1fl/fl mice had reduced mitochondrial protein levels along with increased LC3 level in mitochondria, and these changes were dependent on FUNDC1. We also observed mitophagosomes in liver sections of DEN-treated FUNDC1fl/fl mice, but not of DEN-treated FUNDC1Δhep mice. We have suggested several lines of evidence to describe the critical role of mitophagy in regulation of inflammasome activation. Mitophagy reduced mtDNA release and subsequently inhibited inflammasome activation as measured by caspase-1 cleavage and matured IL1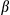 upon treatment of LPS-primed hepatocytes with ATP or poly(dA:dT). Loss of FUNDC1 led to accumulation of dysfunctional mitochondrial and aggravated the activation of inflammasomes. CsA, a blocker of the MPTP, attenuated the inflammasome activation. Elimination of mtDNA by EtBr treatment reduced inflammasome activation in a FUNDC1-dependent manner. It is envisioned that, in response to DEN treatment, mitochondria become damaged, and mtDNA or mitochondrial ROS (mtROS) are released from mitochondria to activate the inflammasome. Mitophagy activation reduces inflammasome activation by eliminating damaged mitochondria. However, chronic inflammation may inhibit mitophagy through a feedback mechanism and amplify mitochondrial damage, leading to excess chronic inflammation.44 DEN-induced cytosolic mitochondrial DNA cannot phosphorylate TBK1 and IRF3 in primary hepatocytes and KCs isolated from 1-month DEN-treated WT and FUNDC1–/– mice, although it is reported that released mtDNA can bind to cyclic GMP-AMP synthase and activates the STING pathway through the TBK1-IRF3 signaling axis.45 Further studies are required to understand how FUNDC1-mediated mitophagy regulates the inflammsome, but not the STING pathway.
upon treatment of LPS-primed hepatocytes with ATP or poly(dA:dT). Loss of FUNDC1 led to accumulation of dysfunctional mitochondrial and aggravated the activation of inflammasomes. CsA, a blocker of the MPTP, attenuated the inflammasome activation. Elimination of mtDNA by EtBr treatment reduced inflammasome activation in a FUNDC1-dependent manner. It is envisioned that, in response to DEN treatment, mitochondria become damaged, and mtDNA or mitochondrial ROS (mtROS) are released from mitochondria to activate the inflammasome. Mitophagy activation reduces inflammasome activation by eliminating damaged mitochondria. However, chronic inflammation may inhibit mitophagy through a feedback mechanism and amplify mitochondrial damage, leading to excess chronic inflammation.44 DEN-induced cytosolic mitochondrial DNA cannot phosphorylate TBK1 and IRF3 in primary hepatocytes and KCs isolated from 1-month DEN-treated WT and FUNDC1–/– mice, although it is reported that released mtDNA can bind to cyclic GMP-AMP synthase and activates the STING pathway through the TBK1-IRF3 signaling axis.45 Further studies are required to understand how FUNDC1-mediated mitophagy regulates the inflammsome, but not the STING pathway.
We have delineated the cascade of events that links FUNDC1-regulated mitophagy with inflammasome activation, thus contributing to tumorigenesis. We have found that activation of caspase-1 was exacerbated in FUNDC1-depleted hepatocytes isolated from liver tissue of DEN-treated mice and in isolated cultured primary hepatocytes. Also, ablation of FUNDC1 resulted in a markedly increased inflammatory response, including the JAK/STAT and NF- B signaling pathways in hepatocytes, leading to hyperproliferation of hepatocytes and tumor initiation (Fig. 7F). Previous reports have shown that IL1β can stimulate IL1R1 in hepatocytes, which up-regulates STAT3 and promotes hepatocellular proliferation.36 We also suggest that IL1β from activated hepatocytes stimulates KCs to secret cytokines, which, in turn, promotes proliferation of hepatocytes. There is growing evidence that inflammasome activation is a major contributor to hepatocellular damage and results in different human and experimental liver diseases, including alcoholic steatohepatitis, chronic hepatitis C virus infection, ischemia-reperfusion injury, and cancer.46 Our study suggests a mechanistic link between mitophagic modulation of inflammatory response and tumorigenesis, and further implies that FUNDC1-mediated mitophagy may potentially represent a therapeutic target during the early stage of tumorigenesis.
B signaling pathways in hepatocytes, leading to hyperproliferation of hepatocytes and tumor initiation (Fig. 7F). Previous reports have shown that IL1β can stimulate IL1R1 in hepatocytes, which up-regulates STAT3 and promotes hepatocellular proliferation.36 We also suggest that IL1β from activated hepatocytes stimulates KCs to secret cytokines, which, in turn, promotes proliferation of hepatocytes. There is growing evidence that inflammasome activation is a major contributor to hepatocellular damage and results in different human and experimental liver diseases, including alcoholic steatohepatitis, chronic hepatitis C virus infection, ischemia-reperfusion injury, and cancer.46 Our study suggests a mechanistic link between mitophagic modulation of inflammatory response and tumorigenesis, and further implies that FUNDC1-mediated mitophagy may potentially represent a therapeutic target during the early stage of tumorigenesis.
Potential conflict of interest
Nothing to report.
Acknowledgment
We sincerely thank Yinzi Ma and Pengyan Xia at Electron Microscopy Laboratory of the Institute of Zoology, Chinese Academy of Sciences, for help with electron microscopy.

