

Nanomedicines for Alzheimer's disease: Therapies based on pathological mechanisms
Abstract
Alzheimer's disease (AD) is the most common neurodegenerative disorder worldwide. Because of the complex pathogenesis of AD and the unique location of AD lesions, effective clinical treatment strategies for this disease remain elusive. However, the development of nanotechnology has allowed a new era of AD treatment to emerge. AD nanomedicines are products of interdisciplinary research that enable high precision and targeted delivery. Additionally, they can specifically regulate various pathogenic factors. This review focuses on nanomedicines based on the pathological mechanisms of AD that can target AD lesions. We also discuss the precise regulatory effects of nanomedicines (including the nanomaterials themselves) on pathogenic proteins, neuroinflammatory molecules, and other pathogenic factors. We summarize the clinical trials that have examined new AD drugs, highlighting the development of new nanomedicines and the progress in their clinical translation. Nanotechnology-based AD treatment is a nascent field, and a complete cure is distant at present; therefore, we also elaborate on the shortcomings of current AD nanomedicines. Finally, we discuss the prospects to guide the future development of AD nanomedicines.
Key points
What is already known about this topic?
-
Alzheimer's disease (AD) drugs face several challenges, such as difficulties in reaching AD lesions and poor potency. Nanotechnology can improve the accumulation of AD drugs in lesions and provide an excellent platform for multi-target treatment.
What does this study add?
-
To elucidate the design principles of anti-AD nanomedicines, this review summarizes the mechanisms by which nanomedicines target and treat AD, highlighting the role of nanomaterials. Some nanomaterials have a photothermal effect that can reduce the abnormal aggregation of pathogenic proteins, and they can scavenge excessive reactive oxygen species to alleviate neuronal apoptosis. In addition, this review summarizes the recent clinical trials of anti-AD drugs to illustrate the status of clinical translation of anti-AD nanomedicines. Finally, we describe the current challenges in this field and propose possible solutions.
1 INTRODUCTION
Alzheimer's disease (AD) is the most common neurodegenerative disorder worldwide. It is characterized by the formation of extracellular senile plaques containing amyloid β (Aβ) peptides and intracellular neurofibrillary tangles (NFTs) containing Tau proteins. The main symptom of AD is severe progressive dementia.1 Imaging methods, such as positron emission tomography (PET) and magnetic resonance imaging (MRI), as well as biomarkers, such as amyloidosis, tauopathy, and neurodegeneration, are used to diagnose and classify AD as preclinical AD, prodromal AD, and dementia due to AD.2 However, an AD diagnosis is often made only after cognitive dysfunction is recognized and reported, either by the patients or those around them. Hence, most AD cases are diagnosed only in the middle or late stages of the disease.3
The early stages of AD involve the progressive loss of cholinergic neurons; therefore, memory can be improved at these stages by upregulating cholinergic levels. Thus, cholinergic upregulation represents an initial treatment strategy for AD. At present, the clinical drugs used to enhance cholinergic levels are divided into two categories: acetylcholinesterase (AChE) inhibitors and butyrylcholinesterase (BChE) inhibitors (Table 1). Tacrine produces similar effects, but it is no longer used because of its high hepatotoxicity.6 Memantine, a low-affinity N-methyl-D-aspartatereceptor (NMDAR) antagonist, is also commonly used as an anti-AD drug in clinical practice; it may exert its effects by alleviating glutamate-induced excitotoxicity. These drugs increase the concentration of acetylcholine in the brain but do not target pathogenic factors. Therefore, although they can delay disease progression, they cannot cure AD. Hence, AD is currently irreversible7 and can only be alleviated, not cured. This clinical challenge places a huge burden on patients with AD and their families. As the global population ages, the prevalence of AD—as a notorious age-related disease—is expected to increase.
| Drugs | Anti-AD pathway | Molecular structure | Ref. |
|---|---|---|---|
| Galantamine | AChE inhibition |  |
[4] |
| Donepezil | AChE inhibition | 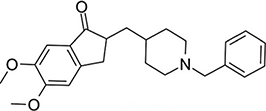 |
[4] |
| Rivastigmine | AChE and BChE inhibition | 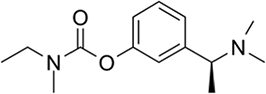 |
[4] |
| Memantine | NMDA-receptor antagonist | 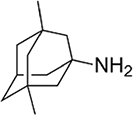 |
[5] |
- Abbreviations: AChE, acetylcholinesterase; AD, Alzheimer's disease; BChE, butyrylcholinesterase.
To address these challenges, researchers have proposed advanced therapeutic strategies for AD in recent years. These strategies include lifestyle changes and disease-modifying therapies targeting Aβ and Tau.8 However, clinical results have shown that these therapeutic strategies have limited efficacy. The main factors hindering their clinical success are (1) the unique physiological structure of the central nervous system (CNS), including the existence of the blood–brain barrier (BBB)9; (2) the extremely complex pathological mechanisms of AD (the exact mechanisms underlying AD onset are unknown, as many factors affect disease progression, and the interactions between these pathogenic factors are unclear10); and (3) the non-regenerative nature of neurons.11 The development or discovery of small-molecule drugs and monoclonal antibodies can overcome these problems. Hence, a delivery system with a strong drug loading and delivery capacity may hold the key to AD treatment.
In the past few decades, the biomedical application of nanotechnology has promoted the rapid development of biosensors and drug delivery systems. Nanomaterials that can enhance the function of biometric components and achieve high signal conversion efficiency have been widely used to develop sensors and probes that can diagnose amyloidosis, tauopathy, and neurodegeneration.12 Nanotechnology has profoundly contributed to the development of AD therapy, and immense efforts have been made to discover new anti-AD drugs to address the shortcomings of current AD treatments. Extracts from traditional Chinese medicines, including curcumin13 and ginkgolide,14 have attracted widespread attention because of their green production processes, low cost, and ability to act on multiple targets. Many studies have demonstrated the anti-AD efficacy of these drugs in vitro. However, their in vivo application is limited by several drawbacks, such as instability, poor solubility, low bioavailability, and a short half-life. Fortunately, nanotechnology can be applied to overcome these drawbacks and promote the clinical translation of these drugs. With the development of precision medicine, targeted nanomedicines have become mainstream. Targeted nanomedicines are valuable because they reduce drug accumulation in normal non-target tissues and increase drug accumulation at lesion sites. Hence, anti-AD drugs approved for clinical use have been incorporated with nanotechnology. For example, converting donepezil into a nanomedicine can extend its half-life and increase its brain-targeting capability. This can enhance the enrichment of donepezil in the brain and reduce drug aggregation in other organs, resulting in improved treatment efficacy and reduced toxicity.15, 16 Accordingly, the application of nanotechnology has moved beyond only drugs with poor characteristics in vivo, expanding to almost all drugs. Moreover, nanoparticles loaded with multiple drugs have made drug co-delivery feasible.17 In addition, researchers are constantly discovering unique biological functions of nanomaterials beyond their role as drug carriers. For example, black phosphorus (BP)-based materials are now known to have antioxidant effects,18 and polyethylene glycol-modified graphene oxide exerts neuroprotective effects by regulating the PIP2-endoplasmic reticulum (ER) stress pathway.19 These biological functions can be leveraged for synergistic AD treatment using nanomaterial–drug combinations. Additionally, most nanomaterials are highly editable and can be modified for targeted action through membrane coating, chemical coupling, and other strategies, enabling targeted delivery and ensuring biocompatibility.20, 21 Therefore, the nanotechnology-based “Trojan Horse” strategy has immense potential to produce AD treatments with low toxicity, high precision, and multi-target regulation.
Nanomedicines have shown great efficacy in the treatment of various diseases. For example, the polymeric micelles called Genexol-PM loaded with poorly soluble paclitaxel has been approved for clinical use in the treatment of lung and breast cancer since 2007.22 In addition, the clinical translation of mRNA lipid nanoparticles has been promoted in recent years to meet the challenge of COVID-19.23 With continuous progress in basic research, the clinical value of anti-AD nanomedicines is also expected to become more apparent in the near future.
Three crucial problems must be addressed to achieve successful AD treatment: (1) drug delivery across the BBB; (2) targeted drug delivery at lesion sites; and (3) regulation of pathogenic factors. We discussed the existing strategies for crossing the BBB in our previous report.24 Thus, this review focuses on lesion targeting and nanomedicine-based strategies for the specific regulation of pathogenic factors. We outline the pathological mechanisms of AD and summarize various lesion-targeting strategies. Numerous previous studies have focused on advanced nanomedicine-based AD therapeutic strategies. Thus, we combined these studies with data on the pathological mechanisms of AD to provide an update on the progress in this field. In addition, we highlight some clinical trials targeting the development of new anti-AD drugs. Finally, we present our views and suggestions for the prospects and directions of this field.
2 AD PATHOPHYSIOLOGY
Despite years of exploration, the pathological mechanisms behind the progression of AD remain controversial. Researchers have proposed several hypotheses regarding AD pathogenesis, which are rooted in distinct core pathological mechanisms such as oxidative stress,25 neuroinflammation,26 Aβ accumulation,10 and Tau accumulation.27 Sufficient evidence supports each of these mechanisms and their role in promoting other pathogenic factors. Hence, it is likely that these pathogenic factors are not independent but instead form a pathological network. Rather than discussing specific hypotheses in detail, we provide a comprehensive overview of the important pathogenic factors associated with AD (Figure 1).

Pathological mechanisms of Alzheimer's disease.
Irrespective of which pathogenic factors cause AD onset, it is certain that neurotoxic Aβ plaques are a key contributor to AD progression. In vitro experiments have shown that Fe3+ binds to the mRNA of the Aβ precursor protein (APP) to promote its expression.28 Under normal physiological conditions, APP is cleaved by α-secretase and β-secretase,29 with Fe3+ playing a role in this reaction.28 Notably, the product generated following cleavage is not pathogenic. However, in some cases, γ-secretase is produced because of mutations of the presenilin (PS) genes (PS1 and PS2). When APP is cleaved by γ-secretase and β-secretase, the product is the Aβ42 monomer, which is prone to abnormal aggregation.30 This process involves Cu2+.31 Interestingly, metal ions play a complex role in AD pathology; while promoting Aβ lesions, metal ions also trigger reactive oxygen species (ROS) generation through the Fenton reaction and induce neuronal apoptosis.32, 33 However, Cu2+ acts as a double-edged sword in AD progression and can help ameliorate AD pathology in the presence of vitamin C.34
Extensive studies have shown that the Aβ42 monomer produces Aβ oligomers that exert neurotoxic effects through multiple pathways. First, Aβ oligomers can activate NMDARs on neuronal membranes and induce ER stress by activating phospholipase C.35, 36 This results in Ca2+ influx37 and subsequently triggers neuronal apoptosis. Second, Aβ oligomers induce cell membrane damage. These oligomers exist in a variety of molecular configurations, each with unique pathological functions. For example, β-sheets can penetrate the cell membrane, creating holes in the lipid bilayer, and ring-shaped oligomers adhere to the cell membrane and then cause membrane damage by assembling into fibrils.38 Third, Aβ oligomers induce mitochondrial depolarization, causing mitochondrial stress, which is accompanied by the production of excessive ROS.38 Notably, excessive metal ions in the brain microenvironment are associated with AD pathophysiology and produce ROS through Fenton/Fenton-like reactions while reducing glutathione levels,39 thus aggravating mitochondrial stress. Together, these processes result in the activation of Caspase-3 and the induction of apoptosis. However, the relationship between Aβ and mitochondrial stress is not a simple upstream or downstream one. Recent evidence showed that mitochondrial dysfunction appeared before Aβ plaques were generated, indicating that Aβ only aggravated mitochondrial damage.40 Therefore, some researchers believe that mitochondrial oxidative stress is the core mechanism of AD and have thus proposed the oxidative stress hypothesis. Fourth, Aβ oligomers can directly and indirectly cause endosomal/lysosomal membrane leakage,41, 42 which results in an enzyme outflow to the cytoplasm and promotes neuronal apoptosis. In addition, Aβ oligomers reversibly produce Aβ fibrils, an important component of senile plaques, although recent evidence suggests that Aβ fibrils do not inherently have neurotoxic effects.43 Under normal physiological conditions, the intracellular autophagy system self-monitors and removes abnormally aggregated pathogenic proteins. However, because PS1 and PS2 are involved in regulating lysosomal Ca2+ homeostasis and autophagosome–lysosome fusion,44, 45 mutations in these genes also impair the autophagy pathway. This bolsters the production and accumulation of Aβ oligomers and fibrils, which are subsequently transported outside the cell. These extracellular plaques impair the functioning of other local cells, especially microglia.
Microglia are the resident immune cells of the CNS, and their function is to maintain microenvironmental homeostasis. Extracellular Aβ oligomers and fibers can be phagocytosed and degraded by microglia, and pattern receptors located on the microglial surface can recognize these pathogenic proteins. This triggers the downstream nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway and mediates nucleotide oligomerization domain (NOD)-like receptor thermal protein domain associated protein (NLRP) 3 recruitment in the presence of ROS. NLRP3 further binds to apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC) and pro-Caspase-1 to generate the NLRP3 inflammasome.26, 46 Under the joint action of NF-κB and the NLRP3 inflammasome, microglia release pro-inflammatory factors that induce neuroinflammation. Notably, these inflammatory factors also promote disease progression via two mechanisms. First, they reversibly promote β-secretase expression and increase Aβ42 production.47 Second, they promote the release of prostaglandin E2, which leads to the release of glutamate, ultimately activating NMDAR and inducing excitotoxicity.26
AD is characterized by abnormal Tau protein accumulation and is thus considered a tauopathy. The physiological function of Tau is regulated by post-translational modifications (PTMs), including phosphorylation, acetylation, glycosylation, and glycosylation.27 Under normal physiological conditions, Tau binds to tubulin to maintain cytoskeletal homeostasis. However, in AD, Zn2+ and Fe2+ induce abnormal PTMs, and Tau is hyperphosphorylated.48 As a result, its binding affinity to tubulin is weakened. Moreover, hyperphosphorylation increases the self-binding affinity of Tau, leading to the formation of Tau oligomers and more advanced NFTs.30 The abnormal behavior of Tau induces cytoskeletal changes, synaptic function impairment, and ER stress. It also physically hinders the axonal transport mediated by dynein and kinesin.1, 49 In addition, hyperphosphorylated Tau (p-Tau) impairs the autophagosome–lysosome system,50 causing the aggregation of pathogenic Tau and the diffusion of abnormal proteins to the extracellular region via exosome- and prion-like propagation.51, 52 Interestingly, Tau-related pathology is closely related to Aβ pathology. Recent evidence suggested that Aβ can promote Tau hyperphosphorylation through the activation of glycogen synthase kinase-3 beta (GSK-3β),53 and in turn, p-Tau can promote Aβ cleavage.54 Recent studies have also suggested a more complex association between tauopathy and microglia. The Tau isoforms Tau 410 and Tau 441 can bind to polyglutamine binding protein 1 (PQBP1) and subsequently activate the cyclic guanosine monophosphate-adenosine monophosphate (GMP-AMP) synthase (cGAS)–stimulator of interferon genes (STING) pathway, eventually up-regulating NF-κB and promoting the release of inflammatory cytokines.55 Conversely, tauopathy can be alleviated by blocking the NLRP3 inflammasome pathway in the microglia,56 although the exact mechanism is unclear.
The current evidence indicates that the pathological mechanism of AD is very complex and involves Aβ fibrils, neuroinflammation, Tau accumulation, oxidative stress, metal ions, autophagy, and other factors. However, this evidence is insufficient to delineate the primary factors that cause AD onset and the core targets of AD progression, making the clinical diagnosis and treatment of AD extremely difficult. Hence, further research on the pathological mechanisms of AD is urgently needed.
3 NANOMEDICINES FOR TARGETED DRUG DELIVERY
The curative efficacy of a drug dependents on more than just its potency. Drug accumulation at the lesion site is a key factor affecting drug efficacy. Therefore, when assessing AD treatment, it is necessary to consider drug concentrations at lesion sites and the effects of the drug on specific pathogenic molecules rather than the whole brain. Nanomedicines have provided two solutions to this problem. First, the pathological features of AD and external stimuli can be used as a gated switch to stimulate drug release, thus enabling responsive release and targeted delivery to AD lesions. Second, a precise drug delivery system can be designed to target affected cells and pathogenic mechanisms using specific biomolecules (Figure 2).

Nanomedicines for targeted delivery to AD lesions. Nanomedicines with long circulation times and blood–brain barrier-crossing abilities adopt two targeting strategies after entering the brain parenchyma. The first involves targeting the AD microenvironment. The AD microenvironment has pathological characteristics such as high ROS levels, the accumulation of pathogenic proteins, and hypoxia, which can act as intrinsic stimuli for nanocarriers. In addition, external stimuli, such as light and ultrasound, can be applied to the lesion site to promote the disintegration of nanocarriers. The released drugs can then be taken up by the surrounding cells and exert their anti-AD effect. The second strategy involves more precise cellular targeting. In this strategy, the high expression of receptors and transporters on the cell membrane of diseased cells is used to achieve the specific intracellular internalization of the nanocarrier, followed by its disintegration inside the cell and subsequent drug release. The second strategy can also enable the further targeted treatment of intracellular pathogenic factors. AD, Alzheimer's disease; ROS, reactive oxygen species.
Several responsive nanomaterials have been developed that enable the fabrication of responsive nanoplatforms with gated properties. The nano-systems can be modified through covalent linkage or physical adsorption strategies.57 Depending on the material, the nanoplatforms may respond to bio-endogenous stimuli, such as ROS,58 pH,59 and hypoxia.60 A variety of responsive material-based nanoplatforms have been developed, including LEGO-like nanoplatforms based on cyclodextrin, cucurbit, and other supramolecules that release the encapsulated drug in response to certain disease-causing proteins.61 Some metal–organic frameworks (MOFs) can release drugs in response to acidic conditions following the disruption of covalent bonds.62 Pathological changes in AD, such as the excess accumulation of metal ions,63 ROS,64 and Aβ,65 serve as excellent environmental stimuli for responsive nanoplatforms. Moreover, these responsive systems promise intelligent drug delivery based on dynamic changes in the pathological microenvironment, preventing overtreatment-induced disruptions to microenvironmental homeostasis. AD pathology affects several cell types, and some pathogenic factors can accumulate and multiply in the extracellular space; therefore, drug release at the lesion site enables the regulation of the entire pathological microenvironment.
The generation of neurotoxic substances and neuroinflammation is believed to induce the loss of cholinergic neurons. Hence, direct and precise drug delivery to affected neurons or activated immune cells is another effective therapeutic strategy. Traditionally, this goal is achieved through the surface modification of nanoplatforms using ligands that specifically recognize or bind to receptors or transporters on the membrane of diseased cells66 or sub-cellular pathological units.67 Moreover, although the interactions between nanomaterials and various proteins have been widely studied, it is still unclear whether the affinity between nanomaterials and membrane-expressed proteins can enable targeted delivery. Recent studies have shown that bundled single-walled carbon nanotubes (bSWNTs) can act as artificial neurotrophins. Upon specific recognition by Trk receptors, they can regulate downstream pathways to improve learning and memory,68 demonstrating that unmodified nanomaterials can still enable targeted delivery. Although several studies have confirmed the feasibility and effectiveness of these approaches, some challenges remain. These include (1) the elimination of the nanomedicines by the immune system during systemic circulation; (2) the interaction between the targeting ligand and serum proteins, which results in the formation of a protein corona complex and a loss of targeting capability69; and (3) the non-specific expression of several target receptors and transport proteins, which results in drug accumulation within non-target tissues. Fortunately, these challenges are not insurmountable. In recent years, researchers have developed biomimetic drug delivery systems that use cell membranes,70 extracellular vesicles,71 and living cells72, 73 as drug carriers. These systems can evade immune clearance because of their endogenous origin, and they exhibit certain characteristics of the parent cell because of the presence of membrane-expressed proteins. For example, nanomedicines based on regulatory T cells (Treg) can exert immunosuppressive effects through immune checkpoints such as cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) and lymphocyte activation gene-3 (LAG-3).74 In addition, after drug loading, extracellular vesicles and cellular carriers remain active and can still perform their normal physiological functions. Therefore, biomimetic drug delivery systems can significantly reduce the off-target effects of nanomedicines and facilitate the synergistic treatment of AD. Although researchers have not yet discovered an endogenous biological material that can target cholinergic neurons, these agents can still be engineered to achieve neuron-targeting effects through lipid insertion, membrane fusion, and genetic engineering.70, 75
After entering lesioned cells, nanomedicines are distributed to subcellular organelles. Previous studies have focused on the targeted delivery of nanomedicines to subcellular organelles, such as the mitochondria,76, 77 Golgi apparatus,78 and ER,79 because the mitochondria are the main site of aerobic metabolism and the Golgi apparatus and ER are closely related to protein expression. Restoring or modifying the function of these subcellular organelles is essential for reversing disease progression. In addition, recent studies have shown that pathological markers generated due to abnormal protein folding, such as Lewy bodies80 and NFTs,81 also act as special subcellular organelles formed by liquid–liquid phase separation. Therefore, targeted drug delivery to these pathological markers is of great interest. Although few studies have targeted subcellular organelles for AD nanotherapy, this approach may represent the next step of precision medicine in AD and warrants more attention.
Table 2 summarizes several recent relevant studies on AD nanotherapeutics. In general, the most popular strategies involve crossing the BBB using targeted nanomedicines and special administration routes, followed by stimuli-response drug release in the pathological microenvironment or direct drug internalization by diseased cells. However, because the core pathological mechanism of AD is unknown, precise delivery to some pathogenic factors does not always improve efficacy. Thus, there is a greater focus on more effective and targeted modification of multiple pathogenic factors via cocktail therapy. Irrespective of whether anti-AD nanomedicines achieve triple-cascade targeting (lesion sites, diseased cells, and pathological factors), they must ultimately improve neurotransmitter release by preventing neuronal apoptosis, relieving neuroinflammation, and promoting nerve regeneration. In the following sections, we elaborate on the applications of nanomedicines for AD therapy.
| Nanoplatform | Cargo | Functional modules for targeted delivery | Intervention target | Ref. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crossing the BBB | Responding to lesion characteristics | Identifying diseased cells | Targeting pathogenic factors | Aβ | Tau | Oxidative stress | Metal ion | Inflammation | Synapses | Others | |||
| Nanoparticle | NAPVSIPQ | TGN | - | Tet-1 peptide | - | √ | √ | √ | - | - | - | [82] | |
| Nanoparticle | Hydrogen | - | - | - | - | √ | - | √ | - | √ | - | [83] | |
| Nanoparticle | Ceria–gold nanorods | Photothermal effect | Reactive oxygen species (ROS)-responsive | - | - | √ | - | √ | - | - | - | [84] | |
| Nanoparticle | Polydopamine, rhein | KLVFFAED peptide | - | - | KLVFFAED peptide | √ | - | √ | - | - | - | [85] | |
| Nanoparticle | Fingolimod, siSTAT3, ZnO | 4-aminophenyl a-mannopyranoside | - | - | - | √ | - | - | - | √ | - | Production of brain-derived neurotrophic factor (BDNF) | [86] |
| Nanoparticle | Nec-1s | CRTIGPSVC peptide | ROS-responsive | - | - | √ | - | √ | - | √ | √ | - | [87] |
| Nanoparticle | Epigallocatechin-3-gallate | Tight junction opening | - | - | - | √ | - | - | - | - | √ | - | [88] |
| Nanoparticle | Aβ peptide, rapamycin | - | - | - | - | √ | - | - | - | √ | - | Nanovaccine for activating Aβ-specific T cells | [89] |
| Nanoparticle | - | - | Near-infrared (NIR) light-responsive | - | - | √ | - | √ | √ | √ | - | - | [90] |
| Nanoparticle | Curcumin, siBACE1 | Phosphatidic acid-functionalized high-density lipoprotein (pHDL) | - | - | pHDL | √ | - | - | - | √ | √ | - | [91] |
| Nanoparticle | TREM2-encoding plasmid | P2 peptide | - | - | - | √ | √ | - | - | √ | √ | - | [92] |
| Nanoparticle | Gadolinium (III), Prussian blue | Angiopep-2 | √ | - | √ | - | √ | - | - | [93] | |||
| Nanoparticle | - | - | - | - | Aβ antibody | √ | - | √ | - | - | - | Decreasing Aβlevels in the blood | [94] |
| Nanoparticle | siBACE1 | Galactose | - | - | - | √ | √ | - | - | - | - | - | [95] |
| Nanoparticle | - | Glutathione | - | - | - | √ | - | - | - | - | - | - | [96] |
| Nanoparticle | Nerve growth factor | Photothermal effect | Thermoresponsive | - | - | - | √ | √ | - | √ | √ | - | [97] |
| Nanoparticle | C3N4 nanodots | Photothermal effect | - | - | Benzothiazole aniline (BTA) | √ | - | √ | √ | - | - | - | [98] |
| Nanoparticle | Clioquinol, donepezil | Transcriptional activator protein | - | - | Monosialotetrahexosylganglioside (GM1) | √ | - | - | √ | - | √ | AChE inhibition | [16] |
| Nanoparticle | Graphitic N dopants | KLVFFAED peptide | - | - | KLVFFAED peptide | √ | - | √ | - | - | - | Photothermal therapy for Aβ disaggregation | [99] |
| Nanoparticle | siBACE1 | T7 peptide | - | Tet-1 peptide | - | √ | - | - | - | - | - | - | [100] |
| Nanoparticle | Ceria nanoparticles | - | - | - | AT8-pTau | - | √ | - | - | √ | - | - | [101] |
| Nanoparticle | - | - | - | - | GGGKLVFF peptide | √ | - | - | - | - | - | Decreasing Aβ levels in the blood | [102] |
| Nanoparticle | Cas9-activating vector | - | - | - | - | √ | - | - | - | - | - | Cas9 for activating Adam10 | [103] |
| Nanoparticle | - | Photothermal effect | Light-responsive | - | - | √ | - | √ | - | - | - | Photothermal therapy for Aβ disaggregation | [104] |
| Nanoparticle | Methylene blue | Angiopep-2 | - | - | Apolipoprotein A-I | √ | √ | - | - | - | - | - | [105] |
| Nanoparticle | Dihydromyricetin | Tg peptide | - | - | - | √ | - | - | - | √ | - | Improving the microbiota–gut inflammasome–brain axis | [106] |
| Nanoparticle | Resveratrol | TGN peptide | - | - | - | √ | - | √ | - | √ | - | Regulating the gut microbiota | [107] |
| Nanoparticle | CRISPR/Cas9 | Glucose, rabies virus polypeptide (RVG) | - | RVG | √ | - | - | - | - | - | CRISPR/Cas9 for editing APP | [108] | |
| Nanoparticle | - | PMAA-PS 80-g-St | ROS-responsive | - | Aβ antibody | √ | - | √ | - | √ | - | Reducing lymphocyte infiltration and re-constructing vessels | [109] |
| Nanoparticle | HNSS peptide | FGL peptide | - | FGL peptide | SS31 | √ | √ | √ | - | - | - | - | [110] |
| Nanoparticle | 6-amino-2-naphthalenesulfonic acid | Biotin | - | - | - | √ | - | - | - | √ | - | - | [111] |
| Nanoparticle | Metformin, donepezil | - | Aβ-responsive | - | - | √ | - | - | - | - | - | - | [65] |
| Nanoparticle | Ag2S quantum dots, siSOX9, retinoic acid | - | - | - | - | √ | - | - | - | - | √ | Promoting neuronal differentiation via siSOX9 | [112] |
| Nanoparticle | Ca2+, folic acid | - | - | - | - | - | - | - | - | - | - | Promoting cholinergic levels in neurons | [113] |
| Nanoparticle | - | - | - | - | - | √ | √ | - | - | - | √ | NIR-mediated differentiation of neural stem cells (NSCs) to neurons | [114] |
| Nanoparticle | Single-stranded DNA | - | - | - | - | √ | √ | - | - | - | √ | NIR-mediated differentiation of NSCs to neurons | [115] |
| Nanoparticle | Hollow manganese prussian white | - | ROS-responsive | - | - | - | √ | √ | - | √ | - | - | [116] |
| Nanoparticle | Prussian blue | CKLVFFAED peptide | ROS-responsive | - | CKLVFFAED peptid | √ | - | √ | - | - | - | Photothermal therapy for Aβ disaggregation | [117] |
| Nanoparticle | - | - | - | - | - | - | - | - | - | √ | √ | Enhancing retention of mesenchymal stem cells and stimulating therapeutic molecule secretion | [118] |
| Nanoparticle | Rutin, siRNA | LRP1, RVG29 | - | RVG29 | - | √ | √ | √ | - | √ | √ | - | [119] |
| Biomimetic nanoparticle | Genistein | Macrophage membrane/RVG29 | - | RVG29 | Triphenylphosphonium (TPP) | √ | - | √ | - | - | - | [120] | |
| Biomimetic nanoparticle | Bexarotene, AgAuSe quantum dots | RVG29 | - | Neural stem cell membrane/RVG29 | - | √ | - | - | - | - | - | - | [121] |
| Biomimetic nanoparticle | Aβ aptamers, CD22shRNA plasmid | Transferrin receptor aptamers | - | - | Aβ aptamers | √ | - | - | - | √ | - | - | [122] |
| Biomimetic nanoparticle | Oxytocin | Macrophage membrane | - | - | - | √ | - | - | - | √ | √ | - | [123] |
| Biomimetic nanoparticle | Flavin mononucleotide | BV2 cell membrane camouflage | - | BV2 cell membrane camouflage | - | - | - | - | - | √ | √ | - | [124] |
| Biomimetic nanozyme | CuxO | - | - | - | KLVFF peptide | √ | - | - | - | - | - | Decreasing Aβ levels in the blood | [125] |
| Extracellular vesicles | - | Inflammatory chemotaxis | - | - | - | √ | - | √ | - | √ | √ | - | [126] |
| Extracellular vesicle | - | N/A | - | - | - | √ | - | - | - | - | - | Improving mitochondrial energy metabolism | [127] |
| Extracellular vesicles | - | Intranasal administration, inflammatory chemotaxis | - | - | - | √ | - | - | - | √ | √ | - | [128] |
| Extracellular vesicles | - | Intranasal administration | - | - | - | √ | - | - | - | - | √ | Enhancing BDNF expression | [129] |
| Micelle | Resveratrol | Neural cell adhesion molecule mimetic peptide C3 | - | C3 peptide | TPP | √ | √ | √ | - | √ | - | [130] | |
| Micelle | Curcumin | Aβ peptide | ROS-responsive | Aβ peptide | - | √ | - | √ | - | - | - | [131] | |
| Micelle | Cathepsin B, siNF-κB | Cyclic peptide DAG | Metal ion-responsive | Tet-1 peptide | - | √ | √ | √ | √ | - | - | [63] | |
| Micelle | Rapamycin | TGN peptide | ROS-responsive | Tet-1 peptide | - | √ | √ | - | - | √ | √ | - | [132] |
| Micelle | - | - | - | - | VQIINK peptide | - | √ | - | - | √ | - | Hyperphosphorylated tau (p-Tau)-targeted nanochaperone | [133] |
| Nanosheet | - | - | - | - | - | √ | - | - | - | - | - | Dissociation of Aβ fibrils by sonodynamic therapy | [134] |
| Nanosheet | - | Photothermal effect | - | - | - | √ | - | √ | √ | - | - | [135] | |
| Nanosheet | - | - | - | - | BTA | √ | - | - | - | - | - | Decreasing Aβ levels via photodynamic therapy | [136] |
| Nanorod | - | COG1410 | - | - | KLVFFA peptide | √ | - | - | - | - | - | - | [137] |
| Quantum dot | Selenium | - | - | - | - | √ | - | √ | - | - | - | [138] | |
| Quantum dot | - | - | - | - | TPP | - | - | √ | - | √ | - | - | [139] |
| MOF | Ceria nanozyme, siSOX9, retinoic acid | - | H2O2-responsive | - | - | - | √ | √ | Promoting the differentiation of NSCs to neurons via siSOX9 | [64] | |||
| Covalent organic frameworks | Superoxide dismutase, catalase | KLVFF peptide | - | - | KLVFF peptide | √ | - | √ | √ | - | - | [140] | |
| Tetrahedral framework | Nucleic acid | - | - | - | √ | - | √ | - | - | - | [141] | ||
| Liposome | Magnesium, siRNA | Matrix metalloproteinase 9 activatable cell-penetrating peptide | - | - | - | - | - | - | - | - | - | [142] | |
| Liposome | Mg2+, CypD siRNA | Matrix metalloproteinase 9 activatable cell-penetrating peptide | - | - | - | √ | - | √ | - | √ | - | Remodeling neurovascular unit | [142] |
| Liposome | Donepezil | Reconstituted HDL | - | - | Reconstituted HDL | √ | - | - | - | - | - | AChE inhibition | [15] |
| Nanogel | Oxytocin | Angiopep-2 | - | - | - | √ | - | - | - | √ | - | - | [143] |
| Nanoparticle cluster | Rapamycin | RVG29 | ROS-responsive | - | - | √ | - | - | - | √ | - | Improvement in the AD brain immune microenvironment | [144] |
| Lipoprotein-like nanostructure | Apolipoprotein E3 | RAP peptide | - | - | - | √ | - | - | - | √ | - | Remodeling of neurovascular units | [145] |
| Nanoflowers | Micro-RNA-124, rutin | RVG29 | - | RVG29 | - | √ | - | √ | - | √ | - | - | [146] |
| Nanoscaffolds | Memantine | - | - | - | - | - | - | - | - | - | - | AChE, BChE, and β-secretase inhibition | [147] |
4 NANOMEDICINES FOR TARGETING TAUOPATHY
p-Tau is closely linked to synaptic dysfunction and is considered a key target for AD treatment and an important biomarker for AD diagnosis. However, p-Tau is very similar to Tau, which is involved in normal physiological functions. Decades of research have only produced a few molecules and short peptides that specifically recognize p-Tau. AT8, an anti-Tau monoclonal antibody extracted from mice immunized with native p-Tau paired helical filament (PHF), can bind to Tau phosphorylated at two or more of the following phosphorylation sites: pS202, Ps202, pT205, and pS208. The affinity of AT8 for different types of phosphorylated Tau varies; particularly, its affinity for pS202/pT205/pS208 triple-phosphorylated Tau is 30 times that for pS202/pT205 Tau,148 indicating that AT8 is more specific to p-Tau. Hence, it is often used as a targeting ligand for p-Tau.
Another group of targeting agents includes the PET tracer for PHF. Ten years ago, researchers developed a molecular probe named T808 that could specifically recognize PHF. Its high affinity for PHF was attributed to the interaction between its 5H-pyrido[4,3-b]indole group and p-Tau. Furthermore, the F atom present in its side chain enabled radioactivity. Hence, this probe could be used for PET imaging.149 T808 was further optimized to produce T807, which had superior biofate and BBB permeability but retained the original targeting group and F atom. Today, T807 is also commonly used as a p-Tau-targeting ligand.150 In addition, the Tau hexapeptide VQIINK can bind to Tau aggregates through hydrogen bonds and hydrophobic interactions and is an important driver of Tau aggregation and propagation.151 Thus, it can also be used as a specific ligand to enhance the recognition of p-Tau.
The clearance of p-Tau is mainly dependent on the self-monitoring function of the CNS as well as upstream PTM regulation. With the discovery of targeted ligands, researchers have proposed the use of nanochaperones, which have a higher specific surface area and capture ability than molecular chaperones, to inhibit tauopathy. The endocytosis of nanomedicines occurs via the following pathways: clathrin-dependent, caveolin-dependent, and clathrin/caveolin-independent endocytosis, macropinocytosis, and membrane fusion. Most nanomedicines enter the endosomal lysosome system through transporters and are rapidly inactivated.152 Therefore, nanochaperones must also be able to escape lysosomes. Leveraging the low pH observed in lysosomes, a recent study used a pH-responsive poly(β-amino ester) to develop nanochaperones that could transform hydrophobic microdomains into positively charged hydrophilic chains under acidic conditions, enabling lysosomal escape. These nanochaperones effectively inhibited the aggregation of p-Tau (Figure 3A,B).133 Paradoxically, the final intracellular degradation of p-Tau–nanochaperone complexes is also dependent on lysosomes. To address this issue, autophagy inducers can be incorporated into the core of the nanochaperone. In another important study, ceria nanoparticles were encapsulated within mesoporous silicon nanoparticles to act as autophagy inducers. The particles were subsequently modified with PEG and AT8 to construct a p-Tau nanochaperone (THN). This nanochaperone inhibited mammalian target of rapamycin (mTOR) and activated the transcription factor EB (TFEB), thereby activating the autophagy/lysosome pathway and clearing any retained intracellular p-Tau-nanochaperone complexes (Figure 3C).101 Interestingly, small molecules such as hydrogen sulfide153 and methylene blue (MB)154 can also alleviate tauopathy by regulating PTMs. However, most of them cannot cross the BBB and must be converted into nanomedicines to achieve in vivo efficacy. In one study, MB was encapsulated in a hydrophilic nanoparticle core, and the particles were surface-modified with BBB-targeting peptides to obtain lipid nanocomposite (APLN)/MB (Figure 3D). These nanoparticles retained the MB-induced up-regulation of phosphorylated protein kinase B (p-Akt) and p-GSK-3β and thus significantly decreased intracellular p-Tau levels and alleviated the neuroapoptosis caused by okadaic acid (Figure 3E–H).105

Nanomedicines targeting tauopathy. (A) Schematic illustration of the Tau nanochaperone (Tau-nChap) selectively capturing intracellular pathological Tau and inhibiting its neuronal aggregation. (B) Transmission electron microscopy images of pathological Tau after incubation with the VQIINK peptide, Tau-VQINK-decorated pegylated micelle, and Tau-nChap at 37°C for 7 days. Scale bar = 200 nm. Reproduced from Ref. [133] with permission. (C) Fabrication of the THN and schematic illustration showing that THN activates the autophagy-mediated clearance of pathogenic tau in AD. THN is administered via intracerebroventricular injection and can enter the hippocampus. THN can simultaneously target and bind to intracellular p-Tau and/or p-Tau paired helical filaments (PHFs) and selectively accumulate in tauopathy-affected cells and brain regions. THN can also enhance autophagic flux by concurrently inhibiting mTOR and activating TFEB, promoting the efficient clearance of pathogenic Tau and restoring Tau homeostasis in the brain. Reproduced from Ref. [101] with permission. (D) Schematic diagram of APLN/MB preparation. (E) Schematic of the mechanisms underlying the inhibition of Tau phosphorylation via the Akt/GSK-3β pathway. (F) Fluorescence images of Tau protein aggregation obtained using the Thioflavin T (ThT) probe. Scale bar = 100 μm. (G,H) Morphological images (G) and apoptosis analysis (H) of okadaic acid (40 nmol/L)-induced tauopathy in SH-SY5Y cells after treatment with MB, PLN/MB, and APLN/MB, respectively. Scale bar = 50 μm. Reproduced from Ref. [105]. (I, J) Schematic illustration of LRsAR preparation (I) and its anti-AD mechanisms (J). (K) GSK-3β mRNA levels in the brain's cortex and hippocampus in Tau.P301S AD mice. Reproduced from Ref. [119] with permission. AD, Alzheimer's disease; THN, p-Tau nanochaperone.
Another strategy to inhibit p-Tau production is gene therapy based on small interfering RNA (siRNA), which can silence target genes with high specificity. However, nucleic acid drugs typically have poor biofate owing to enzymatic hydrolysis and immune clearance. Hence, it is necessary to identify suitable vectors for their delivery. Fortunately, nanocarriers can provide an enclosed environment for nucleic acid drugs, preventing their inactivation in the systemic circulation. Moreover, nanocarriers can enhance their targeting abilities. Therefore, nanocarrier-based delivery systems for nucleic acid drugs have been rapidly developed in the past few years.155 For example, the gene drug LRsGAR was prepared by loading siGSK-3β and bovine serum albumin into two-dimensional magnesium/aluminum nanoparticles modified with targeting peptides (Figure 3I). Intravenous LRsGAR injection significantly decreases GSK-3β mRNA levels in the hippocampus and cortex of Tau.P301S AD mice, and ultimately decreases p-Tau accumulation in the brain (Figure 3J,K).119
In summary, several therapeutic strategies can attenuate tauopathy, including methods that reduce p-Tau expression, clear p-Tau, inhibit p-Tau aggregation and stabilize the cytoskeleton. Nanomedicines allow the precise adoption of the first three therapeutic strategies. However, most studies have focused on neuronal death and have not evaluated the cytoskeleton, even though the cytoskeleton is an important biological factor, and tauopathy causes cytoskeletal changes that lead to neuronal dysfunction even before neuronal death occurs. For example, 5 μM acrolein does not cause neuronal death but triggers synaptic dysfunction.156 Therefore, more research is needed on neuronal cytoskeletons in AD models.
5 NANOMEDICINES TARGETING PATHOLOGICAL Aβ
As a pathogenic protein implicated in AD, Aβ has attracted widespread attention over the past few decades. The key hypotheses of AD pathogenesis are currently centered around the Aβ cascade. Current Aβ-targeted therapies share several similarities with p-Tau-targeted strategies. For example, they can improve the clearance rate of the pathogenic protein by activating autophagy pathways. Therefore, to avoid repetition, we mainly discuss nano-clearance strategies unique to Aβ in this section.
Nature serves as a major inspiration for the discovery of ligands targeting pathological Aβ. Given that pathological Aβ monomers tend to interact and form oligomers, short peptides derived from pathological Aβ (KLVFFAED peptide,131 KLVFF peptide,125 and GGGKLVFF peptide102) have been developed to mimic the aggregation behavior of pathological Aβ. Studies exploring the molecular mechanisms of pathological Aβ have identified special biomolecules closely related to Aβ aggregation. For example, GM1 ganglioside was found to interact with Aβ to form GM1 ganglioside-bound Aβ, which had a unique molecular structure and accelerated Aβ assembly.157 Moreover, high-density lipoprotein (HDL) was linked to Aβ-related pathological mechanisms. In particular, the apolipoprotein A-I-rich HDL subtype was found to exert neuroprotective effects because of its high affinity to Aβ and its effect on Aβ clearance.105 As a result, GM1 and HDL have become common Aβ-targeting ligands. In addition, several synthetic compounds have also shown outstanding Aβ recognition capabilities. Notably, Thioflavin-T fluorescein and its derivatives158 are known to bind to the long axial side chain of Aβ fibrils and are often used to label Aβ fibrils to study fibrosis mechanisms.159 Therefore, modifying the surfaces of nanomedicines with these ligands could greatly enhance their affinity to pathological Aβ, thereby trapping pathological Aβ molecules for further clearance.
Several AD treatment strategies based on the origin and progression of pathogenic Aβ have been proposed. These include inhibiting the production of pathological Aβ monomers, eliminating pathogenic Aβ monomers, preventing Aβ aggregation, and destroying Aβ fibrils. siRNA-based gene therapy has also been applied for pathogenic Aβ, and β-site cleavage enzyme 1 (BACE1)—a key enzyme involved in abnormal Aβ cleavage—appears to be an ideal target. When siBACE1 is encapsulated into polymeric nanoparticles modified with ligands targeting the BBB, siBACE1 nanoparticles can be delivered to the brain parenchyma. Here, they enter diseased cells via the endocytosis pathway and release siBACE1, precisely blocking the translation of BACE1 mRNA and thereby preventing abnormal APP shearing and pathological Aβ production (Figure 4A).95 Another pathogenic Aβ clearance approach based on gene therapy involves CRISPR-Cas9, a revolutionary technology that can target and permanently edit DNA sequences.160 The CRISPR-Cas9 technique can be used to target pathological Aβ in two ways: altering the DNA sequence corresponding to β/γ-secretase or enhancing α-secretase expression. However, γ-secretase is essential to produce non-pathological Aβ, and the absence of β-secretase may have adverse effects. Hence, it may be preferable enhance α-secretase activity to competitively consume APP and thus reduce the production of pathological Aβ. Notably, a disintegrin and metalloprotease (ADAM) 10, a member of the α-secretase family, is a suitable target for enhancing α-secretase activity. Restoring Adam 10-targeted catalytically deactivated Cas9 activity with a nanocomplex based on the R7L10 peptide and Cas9 activator can help reduce pathological Aβ production (Figure 4B).103 Although gene therapy can address the generation of excess pathological Aβ at its source, with AD progression, the amount of pathological Aβ in the brain gradually increases. Thus, merely reducing Aβ production may be insufficient to achieve therapeutic effects. Hence, it is more critical to prevent the accumulation of pathological Aβ and promote its clearance. Nanochaperones are a suitable tool for achieving this goal. For example, an ultra-small nanoparticle core based on hydrophobic N-vinylcaprolactam (VCL), aromatic N-phenylacrylamide (PAM), and acrylic acid (AAc) was fabricated (VP NPs). The ultra-small nanoparticles were further surface-modified with the ROS-responsive ligand 4-hydroxyphenylboronic acid (HPBA) to generate a nanochaperone with microenvironment-targeting properties. Upon exposure to oxidative stress, the HPBA structure of this nanochaperone was destroyed. This exposed the AAc groups, which produced electrophilic carbon ions that subsequently attacked nucleophilic lysine16 in pathological Aβ, eventually creating a VP-Aβ complex (Figure 4C).144 Interestingly, pathological Aβ can be transported from the CNS to the peripheral blood,161 and the decrease in Aβ in the peripheral blood may in turn promote the efflux of Aβ from the brain. Therefore, it may be possible to clear pathological Aβ by bypassing the BBB. To effectively eliminate Aβ from the peripheral blood, nanomedicines must have a suitable half-life. The most popular long-cycle strategy involves camouflaging nanomedicines within an erythrocyte membrane, which can reduce its immune clearance during systemic circulation because of the expression of a “do not eat me” signal (CD47).162 Therefore, nanochaperones can be prepared with erythrocyte membrane vesicles modified with pathological Aβ-targeting peptides. In a recent report, the KLVFF peptide was added to the surface of erythrocyte membranes via lipid insertion. Furthermore, to prevent the Aβ from damaging the erythrocyte membrane structure and improve system stability, CuxO nanoparticles with nanozyme activity were inserted into the obtained EM-K vesicles. Finally, a nanosponge with a long half-life and Aβ-adsorption functions called CuxO@EM-K was constructed (Figure 4D).125 Although this approach was effective against AD, erythrocyte membranes may sometimes act as a double-edged sword for Aβ nanosponges. This is because although CD47 improves the circulation of nanosponges, it also inhibits the uptake of the Aβ–nanosponge complex by monocytes, affecting the eventual immune clearance of Aβ. The underlying mechanism warrants further exploration and could be a new area of focus in research on Aβ clearance and the development of novel adjuvant therapies against AD.

Nanomedicines for targeting pathogenic Aβ. (A) Fabrication of the glycosylated “triple-interaction” stabilized siRNA nanomedicine (Gal-NP@siRNA) and its mechanism and approach for treating AD pathology in APP/PS1 transgenic mice. Reproduced from Ref. [95] with permission. (B) Schematic representation of the Cas9 activator nanocomplex delivery system (top). A schematic representation of in vivo genome activation with the Cas9 activator nanocomplex for AD therapy (bottom). Reproduced from Ref. [103] with permission. (C) Construction of VP@RVG29. Transmission electron microscopy (TEM) images of Aβ monomers (top) and Aβ fibrils (bottom) after incubation with different nanoparticles. Schematic illustration of VP-Aβ generation. Aβ monomers self-assemble into fibrils, while VP@RVG29 can inhibit Aβ fibrillation, depolymerize Aβ fibrils, and promote their reassembly into VP-Aβ composites. Reproduced from Ref. [144] with permission. (D) (I) Preparation of erythrocyte membrane vesicles through hypotonic treatment. (II) Incubation of erythrocyte membranes with Aβ-targeting molecules (DSPE-PEG-K) for the preparation of EM-K vesicles. (III) Fusion of EM-K vesicles with CuxO nanoparticles (CuxO@EM-K). The resulting CuxO@EM-K can capture Aβ in the blood, and the Aβ bound to CuxO@EM-K is then eliminated by the liver. Subsequently, the clearance of peripheral Aβ facilitates a large efflux of Aβ from the brain into the blood through the sink effect, leading to a reduction in the brain's Aβ burden. Reproduced from Ref. [125] with permission. (E) Under ultrasound stimulation, the change in the local dipole moment in BiOCl nanosheets induces piezoelectric polarization (Ppiezo) and separates electron–hole pairs by generating an internal electric field (IEF). The separated charge carriers trigger piezocatalytic redox reactions involving water and dissolved oxygen molecules. The reactive oxidative species produced oxidize the highly stable, self-assembled Aβ aggregates (e.g., Aβ fibrils and plaques) and disaggregate them to generate denatured fragments. Reproduced from Ref. [134] with permission. (F) Illustration of the inhibition and disaggregation capacity of nanoparticles for Aβ42 fibrillation and the elimination of Aβ42 plaques from the AD mouse brain. Reproduced from Ref. [104] with permission. AD, Alzheimer's disease.
Above, we discuss the biological strategies for clearing pathological Aβ. However, physical strategies have also been developed to promote the de-aggregation of Aβ fibrils. These can be divided into ROS-dependent and local heat-dependent strategies. Because of their high polarity and hydrophilicity, ROS can weaken the hydrophobic and hydrogen bonds in Aβ aggregates and oxidize the amino acid residues of Aβ monomers. Together, these effects can together result in the de-aggregation of Aβ fibrils.134 Thus, ROS-based Aβ fibril-scavenging strategies have been developed. Photodynamic therapy (PDT) is a common ROS generation approach. In PDT, the photosensitizer transitions from a stable ground state to a high-energy singlet state after absorbing photons. It transfers a part of its energy to surrounding oxygen molecules through intersystem crossing to generate singlet oxygen and returns to a stable excited three-line state.163 PDT based on BP nanosheets was able to inhibit the progression of pathological Aβ fibrils.136 However, light—unlike ultrasound waves—has a limited ability to penetrate tissues. Thus, sonodynamic therapy (SDT) based on sonosensitizers appears to be a more promising approach.164 Bismuth oxychloride nanosheets can act as sonosensitizers to produce ROS in response to ultrasonic stimulation (Figure 4E), thereby relieving Aβ deposition in AD brains ex vivo.134
Although research has reported promising results, the inhibition of Aβ aggregation via ROS-dependent pathways remains controversial. The AD microenvironment is high in ROS, and the ROS generated by PDT and SDT is also cytotoxic and could aggravate neuron loss. Therefore, the feasibility of ROS-dependent Aβ clearance in AD treatment must be carefully considered and validated. Hence, local heat-dependent strategies may be more suitable for AD. The photothermal effect refers to the local induction of high temperature in response to NIR irradiation. After absorbing photons, the excited photosensitizer returns to the ground state through the conical intersection route, and this transition is accompanied by heat release, resulting in high local temperatures.165 Current evidence suggests that nanoparticles based on guanidinocalix7 arene (GC5A) and the photothermal material PDPP can capture pathological Aβ fibrils. Moreover, local photothermal therapy (PTT)-induced thermal stimulation can inhibit the structural transformation of pathological Aβ from α-helixes to β-sheets, inhibiting its aggregation and promoting the de-aggregation of existing Aβ fibrils (Figure 4F).104 However, the underlying mechanism remains unclear. In addition, because local thermal effects can reversibly increase BBB permeability,166 mild thermal effects that do not cause brain tissue damage are useful for the treatment of AD.
Aβ-related clinical trials have not been as effective as expected, and confidence in Aβ as a target for AD treatment has been reducing.167 However, it must be emphasized that the pathology of Aβ is very complex. Given the existence of many subtypes and forms of pathogenic Aβ, it is challenging to identify the optimal entry point or target for conducting clinical trials on Aβ monoclonal antibodies. In contrast, nanomedicine-based strategies targeting Aβ are more comprehensive and range from eliminating pathological Aβ sources to removing Aβ fibrils. Hence, the application of these nanotechnology-based tools could help improve the clinical success of Aβ-based anti-AD strategies.
6 NANOMEDICINES FOR RESCUING NEURONAL APOPTOSIS
Neuroapoptosis is a major contributor to neuron loss. Neuroapoptosis is triggered by the upregulation of cleaved Caspase-3/9, which typically occurs in response to mitochondrial dysfunction.168 In previous sections, we discussed nano-clearance strategies for pathologic proteins. Here, we describe ROS- and mitochondria-based anti-AD nanotherapies in detail.
Membrane proteins and receptors can enable neuron-targeted delivery. Receptors and proteins are dynamically expressed on the neuronal membrane, and their expression can change under pathological AD conditions. For instance, the expression of lipoprotein receptor protein (LRP)-1 on neuronal membranes decreases with pathological progression (and possibly with aging).169 Therefore, neuron-targeted delivery can only be achieved using receptors and proteins whose expression is unaltered or upregulated in AD. The receptor for advanced glycation end-products (RAGE) is often upregulated due to Aβ pathology. Thus, Aβ peptides derived from Aβ can be used to achieve neuron-targeted delivery.131 In addition, to target trisialoganglioside (GT1b) receptors,170 neural cell adhesion molecules, and fibroblast growth factor receptor 1 (FGFR1)—which are specifically expressed by neurons and are relatively stable—peptides such as Tet-1170 and C3130 have been developed. In addition, other targets that are highly expressed in various brain cells and have multiple targeting functions have also been considered in nanotechnology-based strategies. Common multifunctional target receptors are the transferrin and cholinergic receptors. The former is expressed in the BBB and neurons and can be targeted by the B6 peptide (CGHKAKGPRK) and transferrin169; the latter is expressed in endothelial cells, neurons, and microglia, and rabies virus glycoprotein 29 (RVG29) and FG loop peptide (FGL)110 are often used as its targeting ligands.146, 171 Accordingly, neuron-targeted nanocarriers can be designed on the basis of the specific binding of the target receptors/proteins to the corresponding ligands.
Because of the interrelationship between ROS and mitochondrial stress, the elimination of excessive ROS is crucial to prevent mitochondrial stress. In the AD microenvironment, Fenton reactions mediated by metal ions, especially cuprous ions, promote ROS production and eliminates glutathione (GSH), aggravating ROS damage.48 Therefore, the delivery of metal-ion chelators is a promising therapeutic strategy and has been explored in clinical trials. However, small-molecule chelators are limited by their biofate and metal-ion chelation capacity and thus often fail to demonstrate satisfactory efficacy in vivo (NCT02292238). By contrast, nanomaterials with the ability to cross the BBB and a stronger metal-ion chelation capacity have shown higher valance ratios. Moreover, some of these nanomaterials also have an enzyme-like activity (“nanozymes”) and can synergistically eliminate excessive ROS. Hence, they are more suitable for AD therapy.172 For example, Nb2C, a two-dimensional nanomaterial with an ultra-high specific surface area, has photothermally mediated BBB-crossing activity and can chelate metal ions (Figure 5A). Nb2C treatment significantly improved mitochondrial function in vivo in an ascorbic acid (AA)- and copper ion-induced neuronal oxidative stress animal model (Figure 5B). Nb2C also showed a strong anti-AD effect in animal studies.135 A groundbreaking study showed that similar two-dimensional nanomaterials, including BP, could cross the BBB and alleviate mitochondrial dysfunction in SH-SY5Y cells by chelating metal ions.18 However, some nanozymes contain copper, iron, and other metals, and their metabolites—especially cuprous and ferrous ions—may reactivate ROS pathological cascades, which could be a matter of concern. However, we speculate that the reactivation of AD progression by metal-containing nanozymes likely depends on their metabolic profile. For example, nanozymes used in AD therapy typically react with ROS and are physiologically stable.174 Hence, they are unlikely to continuously release metal ions after ROS is eliminated from the pathological microenvironment. Instead, they are more likely to be transported to the periphery through the cerebrospinal fluid or other routes.

Nanomedicines for rescuing neuronal apoptosis. (A) Schematic diagram showing the unique functionality of 2D Nb2C MXenzyme with high blood–brain barrier permeability under second near-infrared (NIR-II) irradiation. The MXenzyme captures superfluous Cu2+ and has multiple enzyme-mimicking functions that catalyze ROS scavenging, primarily demonstrating superoxide dismutase (SOD)-, catalase (CAT)-, and peroxidase (POD)-like activities. (B) Representative confocal laser scanning microscopy images of intracellular ROS levels after different treatments. Reproduced from Ref. [135] with permission. (C) Construction of FGL-NP(Cit)/HNSS and acid-responsive features of charge reversal and drug release. (D) Lysosomal escape and mitochondrial targeting of FGL-NP(Cit)/HNSS. (E) Transmission electron micrographs showing the ultrastructure of mitochondria from the hippocampal CA1 region in treated mice. Yellow arrow: damaged mitochondria, white arrow: normal mitochondria. Reproduced from Ref. [110] with permission. (F) Illustration of APBP nanoconjugate preparation. (G) Scheme of the neuroprotection mechanisms of the APBP nanoconjugate. (H) Flow cytometry analysis of cell apoptosis based on Annexin V-FITC/PI staining after various treatments. Reproduced from Ref. [173] with permission. (I) Schematic of the enhanced neuroprotection provided by nanoparticles constructed by Fe-rhein-polydopamine coordination and KLVFFAED peptide (K8@Fe-Rh/Pda NPs) by reversing the “pathological chain of events” in AD. (J) Schematic of the antioxidant mechanisms of K8@Fe-Rh/Pda NPs. Reproduced from Ref. [85] with permission. (K) Neurons (SH-SY5Y cell line) were pretreated with 2 × 10−6 m Aβ1–42 proteins in the absence or presence of extracellular vesicles (EVs) and EVs-SHP2 for 24 h. Colocalization between mitochondria (green, stained with MitoTracker) and lysosomes (eed, stained with LysoTracker) was observed (left). Nuclei were stained with 4′6-diamidino-2-phenylindole (DAPI, blue). Yellow represents colocalized points. Scale bar: 25 μm (left). The colocalization of mitochondria (green, stained with Tom20) with autophagosomes (red, stained with LC3B) was also observed. Nuclei were stained with DAPI (blue). Yellow represents colocalized points. Scale bar: 50 μm (right). (L) SH-SY5Y cells were fixed and examined for autophagosome formation using transmission electron microscopy (left). Scale bar: 5 μm. (M) The JC-1-mitochondrial membrane potential assay was performed using flow cytometry. (N) SH-SY5Y cells were labeled with Annexin V/PI and analyzed by flow cytometry. Reproduced from Ref. [126] with permission.
A more efficient mitochondrial rescue strategy involves the precise delivery of antioxidants; this often requires the assistance of mitochondrial targeting peptides such as TPP.175 In most studies, these mitochondria-targeting peptides have been added to the outer surface of the nanomedicines. However, this approach is not universal because some nanomedicines may disintegrate during lysosome escape and directly release the drug into the cytoplasm, reducing their mitochondrial targeting ability. Linking drugs to mitochondrial targeting peptides via covalent bonds is a promising solution to this problem. For example, the SS31 peptide (which is also an antioxidant and has mitochondria-targeting functions) can be linked to the neuroprotective peptide S14G-Humanin (HNG) to construct a positively charged hybrid peptide (HNSS) polymer that can be encapsulated in citraconic acid (Cit)-modified poly(ethylene glycol)-poly(trimethylene carbonate) polymer (PEG-PTMC). In the prepared NP(Cit)/HNSS nanoparticles, Cit not only provides electronegativity to facilitate HNSS loading but also induces electrical changes during endosome/lysosome transport to promote lysosome escape. In one study, an FGL peptide was added to the surface of NP(Cit)/HNSS, resulting in the construction of a nanoparticle with multiple neuron-mitochondrial targeting functions (Figure 5C). Fluorescein isothiocyanate (FITC) was used to label HNSS, and time-dependent lysosome escape and mitochondrial aggregation were observed (Figure 5D). Furthermore, bio-transmission electron microscopy (TEM) showed that the final nanoparticles significantly reduced the mitochondrial damage caused by AD pathology (Figure 5E).110
In addition to antioxidant delivery, another strategy to eliminate excessive ROS is to stimulate the cells' self-protection mechanisms. Nuclear factor-like 2 (Nrf2) plays an important role in protecting cells from oxidative stress by binding to Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm under physiological conditions. To maintain physiological homeostasis, Nrf2 is activated in cells stimulated by external ROS. Nrf2 can then trans-localize to the nucleus to promote the secretion of downstream antioxidant proteins.176 However, in the AD microenvironment, even though cells are damaged by high ROS levels, Nrf2 maintains abnormally low levels in the cytoplasm. Thus, the activation of the Nrf2 pathway can also serve as an important mode of ROS clearance. To achieve the nuclear transfer of Nrf2, LQLDEETGEFLPIQ peptide (p-Nrf2), a short peptide that competes with Keap1, can be encapsulated in a ROS-responsive phenylboronic dendrimer (a nano-copolymer named APBP) (Figure 5F). In neurons, APBP can prevent the ubiquitin-protease system from clearing Nrf2 and promote its nuclear transport, thus upregulating antioxidant stress proteins (Figure 5G) and protecting the neuron from pathological Aβ-induced apoptosis (Figure 4H).173 Studies on animal models have demonstrated that ROS depletion can alleviate neuronal apoptosis; however, if mitochondria are damaged before excessive ROS is eliminated, they can continue to release cytochrome c and promote apoptosis. Therefore, blocking the pathogenic signals of damaged mitochondria is particularly critical.
Another method to inhibit mitochondria-triggered apoptosis involves direct mitochondrial modification. Oxidative stress can activate mitochondrial fusion, part of a cellular protective mechanism that provides a new membrane to damaged mitochondria to prevent leakage.177 Therefore, stimulating mitochondrial biogenesis is a promising approach for AD treatment. Gao's group attempted to activate mitochondrial biogenesis with a nanomedicine.85 They loaded rhein into a K8-targeting peptide-modified nanocarrier. Upon entering the brain, the nanoparticles released rhein, which exerted antioxidant effects due to its phenolic hydroxyl structure and activated the SIRT1-PGC-1α pathway, upregulating downstream NRF1 expression and promoting mitochondrial biogenesis. The findings suggested that newly generated healthy mitochondria not only secrete antioxidant enzymes to combat intracellular ROS but also reduce and repair damaged mitochondria via mitochondrial fusion (Figure 5I,J). However, some studies have proposed that individual cristae within a specific mitochondrion are functionally independent.178 Hence, the fusion of damaged mitochondria does not always indicate their functional repair. Mitophagy is a major pathway for removing damaged mitochondria during the mitochondrial life cycle. Several drugs that activate mitophagy have been developed, including NAD+ precursors, urolithin A, actinonin, spermidine, rapamycin, and metformin.179 However, few studies have investigated their effects on mitophagy in the context of AD nanotherapy. In a recent study, bone marrow-derived mesenchymal stem cells (MSCs) were transfected with tyrosine phosphatase-2 (SHP-2) using a viral vector. SHP-2 could induce mitophagy and the subsequent generation of SHP-2-overexpressing extracellular vesicles (EVs-SHP2).126 Following the EVs-SHP2 intervention, there was obvious colocalization occurred between lysosomes and mitochondria in pathological neurons (Figure 5L). This phenomenon was attributed to the activation of autophagosomes and the phagocytosis of damaged mitochondria (Figure 5M). Furthermore, mitophagy decreased the green fluorescence of the JC-1 probe and increased red fluorescence, suggesting an improvement in intracellular mitochondrial function (Figure 5N). Hence, neuronal apoptosis was ameliorated (Figure 5O).
In summary, the existing nanomedicine-based strategies for preventing neuronal apoptosis mainly target mitochondria and inhibit downstream Caspase apoptosome activation through ROS scavenging, mitochondrial biogenesis, and mitophagy induction. This strategy provides satisfactory anti-AD efficacy in animal models. However, given the huge difference between animal models and human patients, the clinical value of these strategies needs to be confirmed.
7 NANOMEDICINES FOR ALLEVIATING NEUROINFLAMMATION
Neuroinflammation is a key contributor to AD progression. The pathological network of neuroinflammation is very complex, involving the activation of CNS immune cells (microglia and astrocytes) and the recruitment of peripheral immune cells (lymphocytes, monocytes, neutrophils, etc.).180 The release of pro-inflammatory cytokines induces neuronal pyroptosis. Therefore, in this section, we focus on microglial regulation strategies and non-microglial strategies targeting the amelioration of neuroinflammation.
To suppress neuroinflammation, targeted delivery—which reduces drug immunotoxicity and enhances drug efficacy—is necessary. However, the CNS contains different types of immune cells, and it is often necessary to adjust targeting strategies depending on the target cells. Targeted ligand-mediated delivery is typically directed toward microglia and is primarily dependent on cholinergic receptors expressed on the microglial membrane. The ligands are the same as those used for neurons. Another unique approach involves delivering drugs to inflammatory lesions. Given the characteristics of peripheral immune cell recruitment, these cells or their cell membranes and EVs can be used as drug carriers to transport drugs to inflammatory lesions. However, in biomimetic strategies for brain drug delivery, the timing and location of drug release remain unclear. At present, only one report has suggested that the rapid capture of neutrophils by neutrophil extracellular traps at the inflammatory lesion site results in rapid degradation and the release of loaded drugs.181 Biomimetic drug delivery systems based on macrophage123 and microglia124 membranes can help the drug reach the target lesion and become endocytosed by microglia, but it is still unclear how the drug is released. One recent study showed that a biomimetic carrier based on osteoclast membranes can circumvent lysosomes and achieve drug release through membrane fusion, and the absorbed drug can also be transported between cells via exosomes.182 However, Li et al. reported that the specific uptake of MES23.5 membrane-based nanoparticles by microglia is mediated by the recognition of vascular cell adhesion molecule 1 (VCAM-1) and α4β1 receptors,183 consistent with another simulation study.184 These findings demonstrate the differences in endocytosis pathways between different biomimetic carriers and the possibility of different modes of drug release. One approach to circumvent this problem lies in incorporating a responsive phospholipid component into the cell membrane so that it can disintegrate in the neuroinflammatory microenvironment. When the drug is released, it is considered “foreign” and taken up by immune cells. Then, only lysosome escape is required. In addition, peripheral immune cells can be pre-engineered and programmed via vaccines, and immune organs, such as the lymph nodes and the spleen, can be targeted to modify these peripheral immune cells at their source.
Microglia are the most critical mediators of neuroinflammation, and thus their regulation is crucial. Hence, blocking intracellular pro-inflammatory signaling pathways in the microglia is a current research focus. Given that the microglial activation pathway is involved in the pathological mechanisms of AD, blocking the activation of the NLRP3 inflammasome and the NF-κB pathway is an outstanding strategy. While most recent studies have focused on eliminating pathogenic proteins and ROS to influence the above pathways, other strategies can also be applied. For example, abnormal riboflavin metabolism induced by the deletion of riboflavin kinase (RFK) can activate the tumor necrosis factor receptor-1 (TNFR1)/NF-κB pathway, leading to the excessive activation of microglia. Flavin mononucleotide (FMN), a metabolite of RFK, can inhibit RFK expression by regulating lysine-specific methyltransferase 2B (KMT2B), thereby alleviating microglia-mediated neuroinflammation. However, because of its poor biodistribution, the direct administration of FMN does not improve lipopolysaccharide (LPS)-induced neuroinflammation. To address this challenge, in one study, FMN was encapsulated in human serum protein (HSA) to generate a core that was further coated with a BV2 cell membrane. The resulting biomimetic microglial nanoparticle loaded FMN (MNPs@FMN) could cross the BBB and precisely target the microglia (Figure 6A,B). Water maze test results showed that LPS-induced neuroinflammation led to cognitive dysfunction in mice (Figure 6C), which could be ameliorated by MNPs@FMN treatment. Furthermore, MNPs@FMN treatment also reduced pro-inflammatory factors in the hippocampus (Figure 6D). Immunofluorescence staining showed that after LPS treatment, the endpoint voxels of microglia decreased and their volume increased, indicating that microglia were in an active state. However, the morphology after MNPs@FMN treatment was similar to that in the control group, demonstrating that MNPs@FMN could also inhibit the abnormal activation of microglia in vivo (Figure 6E,F). A similar phenomenon was also observed in astrocytes (Figure 6G,H).124 In addition, recent studies have revealed that mitogen-activated protein kinase (MAPK), IκBα, triggering receptor expressed on myeloid cells 2 (TREM2), and other signaling factors can also regulate NF-κB. The former two can be targeted by small-molecule drugs such as oxytocin143 and rhein,185 whereas TREM2 can mainly be targeted by nucleic acid drugs, such as TREM2-encoding plasmid92 and siRNA.186 By contrast, although NLRP3 inflammasome-mediated neuroinflammation involves multiple steps (NLRP3 production, binding to ASC and Caspase-1, activating GSDMD to form GSDMD-N, and promoting pro-IL-1β maturation) and provides multiple intervention targets,187 research on targeting the NLRP3 inflammasome for AD nanotherapeutics is still limited.

Nanomedicines for alleviating neuroinflammation. (A) Schematic illustration showing the preparation of MNPs@FMN. (B) Schematic diagram showing the proposed mechanism through which MNPs@FMN cross the blood–brain barrier (BBB) and are recruited by microglia. Briefly, MNPs@FMN penetrate the BBB by binding to cell surface receptors on brain endothelial cells. Subsequently, MNPs@FMN accumulate in the microglia, and the released FMN inhibits RFK via KMT2B, while RFK promotes the TNFR1/NF-κB signaling pathway. Ultimately, MNPs@FMN restore cognitive function by suppressing the inflammatory response. (C) Representative path tracks in each quadrant during the probe trial. (D) Protein expression levels of IL-1β, IL-6, and TNF-𝛼 in the hippocampi of lipopolysaccharide (LPS)-treated mice receiving NPs@FMN, MNPs, or MNPs@FMN. (E) Immunofluorescence staining of ionized calcium binding adapter molecule 1 (Iba1)-positive cells in the CA1 region of the hippocampus in LPS-treated mice receiving NPs@FMN, MNPs, or MNPs@FMN. Scale bar: 40 μm. Magnified images are shown in the middle column, and skeletal diagrams of Iba1-positive cells are shown at the bottom of panel G. Scale bar: 10 μm. (F) Quantification of Iba1-positive cells. (G) Immunofluorescence staining for glial fibrillary acidic protein (GFAP)-positive cells in the hippocampi of LPS-treated mice receiving NPs@FMN, MNPs, or MNPs@FMN. Scale bar: 40 μm. Magnified images are shown at the bottom of panel K. Scale bar: 10 μm. (H) Quantification of the numbers and volumes of GFAP-positive cells in the CA1 region of the hippocampus. Reproduced from Ref. [124] with permission. (I) Mode of action and side effects (neuroinflammation) of lipid nanoparticles (LNP)-Aβ, and the proposed therapeutic mechanisms of LNP-R/Aβ for the treatment of Alzheimer's disease (AD). The LNP-Aβ vaccine generates anti-Aβ antibodies but also has adverse effects such as meningitis. By contrast, LNP-R/Aβ can remove Aβ plaques without causing neuroinflammation and provides neuroprotection. Intradermal injection of the LNP-Aβ vaccine leads to the generation of Aβ-specific follicular helper T (Tfh) cells and Th1 cells, resulting in Aβ plaque removal and neuroinflammation. By contrast, the intradermal injection of LNP-R/Aβ leads to the generation of Aβ-specific Tfh cells and Treg cells, resulting in Aβ plaque removal and neuroprotection. Reproduced from Ref. [89] with permission. (J) Spearman's correlation analysis for specific gut microbes and pro-inflammatory cytokines in APP/PS1 mice. The color intensity indicates the level of correlation. *p < 0.05 and **p < 0.01. (K) Effects of different SeNP surface modifications on gut microbiota composition in APP/PS1 mice. (L) NLRP3 (red) immunofluorescence staining in mouse intestinal and hippocampal tissue. Scale bar: 100 μm. Reproduced from Ref. [106] with permission.
Chemokines secreted by the brain mediate the immune infiltration of the brain parenchyma by peripheral-derived neutrophils, monocytes, cytotoxic T cells, helper T cells (Th cells), regulatory T cells (Treg), and B cells. This self-regulatory mechanism serves to clear pathogenic factors. However, these peripheral immune cells have dual pro-inflammatory and anti-inflammatory effects, and hence, their role in AD progression is still controversial.180 It is well-established that cytotoxic T cells and Th cells secrete pro-inflammatory factors, and Treg cells suppress inflammation. Specifically, the membranes of Treg cells express CTLA-4, CD27, LAG3, and other immune checkpoints. These molecules can bind to CD80/86, CD70, and MHC II expressed on the membranes of cytotoxic T cells and macrophages, inhibiting T cell function and inducing macrophage apoptosis.188 This demonstrates the feasibility of two potential Treg-based anti-neuroinflammatory approaches. The first involves constructing Treg membrane vesicles to produce anti-inflammatory effects via membrane proteins. The vesicles can also be used to encapsulate nanoparticles to achieve a synergistic effect between the Treg cell membrane and anti-inflammatory drugs. At present, the anti-inflammatory effect of the Treg membrane has only been verified in other inflammatory models189 and has yet to be applied to neuroinflammation, although it is also expected to be effective in this model. The second approach involves stimulating the proliferation of peripheral Treg cells that are engineered to increase their enrichment and potency in the brain parenchyma. For example, the poor efficacy of the anti-Aβ vaccine is closely related to neuroinflammation, especially because mature dendritic cells (DCs) carrying the Aβ antigen activate Aβ-specific Th cells and promote the secretion of inflammatory factors. Although some of the Aβ can be successfully removed, the high levels of pro-inflammatory factors continue to promote AD pathogenesis. To address this concern, the Aβ vaccine can be co-delivered with rapamycin to obtain tolerant DCs (tDCs) by interfering with the mTOR pathway. The resulting tDCs secrete TGF-β, IL-10, and other anti-inflammatory factors, thereby promoting the production of Aβ-specific Treg cells that can enter the brain parenchyma and exert anti-inflammatory effects (Figure 6I).89
The links between the peripheral system and neuroinflammation are not only mediated by immune cells. Increasing evidence shows that gut dysbiosis and neuroinflammation are interrelated. Specifically, microbial metabolites, cytokines, and misfolded proteins enter the brain via the damaged BBB, lymphatic vessels, and the vagus nerve.190, 191 Studies have shown that the gut microbiota differ significantly between APP/PS1 and healthy mice and is closely related to the levels of pro-inflammatory factors (Figure 6J). However, treatment with chitosan (CS)-selenium nanoparticles (SeNPs) loaded with dihydromyricetin (DMY) can significantly alter the gut microbiota of APP/PS1 mice. Furthermore, after surface modification with the target ligand Tg, the nanodrug can alter the gut microbiota of APP/PS1 mice so that it is similar to that of healthy mice (Figure 6K). In addition, it can decrease NLRP3 expression levels in both the gut and the hippocampus, indicating the simultaneous alleviation of inflammation and neuroinflammation (Figure 6L).106 These findings demonstrate a link between peripheral inflammation and neuroinflammation. However, this area of research is still in its infancy, and more direct evidence is needed to confirm that nanomedicines can regulate neuroinflammation by restoring gut microbial homeostasis.
The immune system is extremely intricate, especially in the CNS, which was initially recognized as a special tissue with immune privilege. Improvements in detection technology allowed the discovery of “invasion” by peripheral immune components (cells and metabolites) in the CNS under pathological conditions. The synergy between these peripheral immune components and CNS regulators can act as a double-edged sword in the context of AD progression. The immune components of the brain are not well understood, but even the known immune components, such as astrocytes, are underutilized. These immune components interact with other pathological factors to build a complex AD immune microenvironment and ultimately promote disease progression. The AD immune microenvironment is considered similar to that of the tumor microenvironment. However, nanomedicines offer an opportunity to escape this immune quagmire through multi-target therapy.
8 NANOMEDICINES FOR STEM CELL THERAPY
It is believed that the neurons in the adult brain cannot regenerate. Hence, even if all pathogenic factors are eliminated by multi-target therapy, only the progression of AD will be halted—the lost neuronal function will not recover. A “cure” is defined as a return to a healthy state; therefore, it is necessary to compensate for the lost neurons by replenishing them. Regenerative medicine provides an excellent platform to achieve this massive goal.
One way to repair injured brain tissue is to grow organoids in vitro and then implant them at the site of injury to replace lost neurons. However, organoids lack blood vessels, and therefore, implanted organoids cannot survive independently in vivo.192 Neurotrophins, such as brain-derived neurotrophic factor (BDNF), are important for promoting neuronal growth. However, because of the presence of misfolded proteins, neuroinflammation, neuronal apoptosis, and other factors in the AD microenvironment, the levels of these cytokines are greatly reduced.193 Thus, the supplementation of these growth factors is very helpful for AD therapy.
In recent years, many studies have focused on delivering growth factors for the treatment of AD via nanocarriers.97, 194, 195 However, it is difficult to accurately control the amount and site of growth factor release. The off-target effects and excessive release of growth factors in peripheral tissues may have adverse effects, such as chronic pain due to excessive nerve growth factors.196 In contrast, the metabolic behavior of living cells is regulated by metabolites from other cells or extracellular vesicles. Hence, delivering living cells with the ability to secrete growth factors is a safer option. MSCs have been widely used in the treatment of various diseases because of their strong immunomodulatory effects. Iron oxide nanoparticles (IONPs) can be internalized by MSCs, where they are degraded into iron ions. These ions can activate the c-Jun N-terminal kinase pathway and upregulate the expression of downstream growth factors, such as BDNF. In addition, IONP-loaded MSCs acquire magnetic properties and can be enriched in the brain parenchyma under an external magnetic field, reducing adverse effects caused by off-target activity (Figure 7A).118 In addition, MSCs and other exogenous stem cells (mainly pluripotent stem cells and neuronal stem cells197) can be transformed into highly differentiated somatic cells. Traditionally, the differentiation behavior of stem cells is regulated by environmental factors, such as cytokines, nutritional status, and mechanical stress.198 However, recent studies have shown that this behavior can also be modulated by nanomaterials. For example, a recent study demonstrated that high levels of Map2 and c-Fos can be induced in neuronal stem cells using circularly polarized light (CPL)-activated L-type NPs (Figure 7B,C).114 Long-term treatment with L-type NPs + CPL in 2xTg AD mice significantly increased the number of neurons in the hippocampus, thereby improving cognitive function (Figure 7D,E). In this study, the neuronal stem cells were not exogenous, circumventing the immune rejection that arises from the injection of undifferentiated exogenous stem cells. In addition, contrary to popular belief, Fu et al. demonstrated that highly differentiated somatic cells can also be reprogrammed into neurons.199 This revolutionary finding provides new avenues for regeneration in neurodegenerative diseases, although it has not yet been applied to AD treatment.

Nanomedicines for regenerative therapy in Alzheimer's disease (AD). (A) Schematic illustration showing the manufacturing of iron oxide nanoparticle-incorporated mesenchymal stem cells (MSC-IONP) for the treatment of AD. Reproduced from Ref. [118] with permission. (B) Illustration of neural stem cell (NSC) differentiation pathways in AD mice induced by chiral nanoparticles (NPs) under 808 nm NIR laser irradiation. (C) Confocal images of NSCs incubated for 7 days under different experimental conditions (left). Red: Map2 for mature neurons. Blue: DAPI for nuclei. Green: GFAP for astrocytes. Scale bar: 100 μm. Confocal images of c-Fos protein expression in NSCs incubated under different experimental conditions (right). Red: c-Fos for active neurons. Blue: DAPI for nuclei. Scale bar: 100 μm. (D) Nissl staining of neuronal cells in the brains (hippocampus) of wild-type (WT) and AD mice from different treatment groups. (E) Track sheets of WT and AD mice after different treatments. Reproduced from Ref. [114] with permission. (F) Schematic of multifunctional NSC therapy for the treatment of AD. (G) Schematic of signaling pathways for NSC differentiation regulated by the PPAR-siSOX9 nanoformulation. (H) Immunofluorescence assay for Aβ at 6 months after treatment. (I) Nissl staining 6 months after treatment. (J) Immunofluorescent detection of Tuj1 6 months after treatment. Reproduced from Ref. [112] with permission. (K) Immunofluorescence staining for CD13 and tomato lectin in the cortex. Scale bar: 20 μm. (L) Immunofluorescence staining for aquaporin 4 (AQP4) and tomato lectin in the cortex. Scale bar: 40 μm. (M) Immunofluorescence co-staining for NeuN (neurons), MAP2 (dendrites), and tomato lectin in the cortex. Scale bar: 20 μm. (N) Escape latency of APP/PS1 mice in the Morris water maze test. Reproduced from Ref. [145] with permission.
Regenerated neurons continue to encounter pathogenic factors, and thus, the clearance of these pathogenic factors is still necessary. To meet the dual demands of neuronal regeneration and pathogenic factor clearance, engineered stem cells can be applied. For example, in one study, neural stem cells (NSCs) were first transfected to overexpress neprilysin (NEP), an Aβ-degrading protease, so that they could clear Aβ via membrane-expressed and exosome-loaded NEP. The engineered NSCs were then induced to phagocytose nanocomposites composed of retinoic acid (RA), Ag2S quantum dots, and siSOX9. RA and siSOX9 are known to regulate the RA and Wnt/β-catenin pathways, respectively, and they synergistically promote the differentiation of NSCs into neurons. In addition, Ag2S quantum dots can produce fluorescence under NIR-II irradiation, assisting the implantation of engineered NSCs (Figure 7F,G). Immunofluorescence and Nissl staining of hippocampal sections confirmed that the engineered NSCs could successfully ameliorate Aβ accumulation and neuronal loss in AD mice (Figure 7H–J).112 However, complex engineering procedures increase the difficulty of fabricating cell preparations, and it is difficult to avoid immune rejection following exogenous stem cell implantation. Thus, avoiding potential immune rejection while adding pathogenic clearance functions to engineered stem cells is a key challenge.
In addition to neuronal loss, AD pathology is accompanied by vascular lesions, including intracranial atherosclerosis, cerebral amyloid angiopathy, BBB damage, and cerebrovascular dysfunction. These effects are caused by a combination of natural aging and pathogenic factors.200 Among them, the dysfunction of the neurovascular unit (composed of endothelial cells, microglia, astrocytes, pericytes, neurons, and other components) causes the influx of neurotoxins from peripheral blood and reduces cerebral hyperperfusion. This results in an insufficient energy supply, affecting metabolite clearance and ultimately promoting AD progression.201 However, remodeling the neurovascular unit is more complicated because it involves the co-regulation of multiple cells. To achieve this goal, the co-delivery of multiple active ingredients is necessary. GM1 is a lipid component that promotes the repair of injured neurons and can self-assemble with DMPC, RAGE-targeted ligand, and apolipoprotein E3 (ApoE3) to form RAP-RL.145 Immunofluorescence imaging of brain slices showed that RAP-RL could improve the function and coverage of endothelial cells, astrocytes, and pericytes in the neurovascular unit (Figure 7K,L) and increase dendrites in hippocampal neurons (Figure 7M), indicating successful neurovascular unit remodeling. In addition, RAP-RL treatment could ameliorate cognitive impairment in AD mice, as demonstrated by the water-maze test (Figure 7N). These findings highlight the effectiveness of neurovascular unit repair, suggesting that vascular repair—like other disease-modifying strategies—could be a valuable component of the therapeutic cocktail for AD.
In summary, at present, the application of stem cell therapy for AD treatment mainly involves growth factor delivery, exogenous stem-cell delivery, endogenous stem-cell induction, and the remodeling of neurovascular units. The focus has shifted from promoting the regeneration of injured neurons to replacing lost neurons without inducing immune rejection. However, most recent studies (including those targeting the regulation of endogenous stem cell differentiation and the reprogramming of functional cells, as well as those exploring the supplementation of exogenous cells) have only focused on neuronal regeneration. Studies involving the remodeling of the entire microenvironment are rare. If the microenvironment is not modified, regenerated neurons will be exposed to the pathological factors causing AD. Thus, it is necessary to promote the regeneration of lost neurons while simultaneously eliminating pathogenic factors. Otherwise, it will only be possible to delay or halt disease progression and not reverse it.
9 STATUS OF CLINICAL TRIALS
Because commercially available drugs cannot cure AD, clinical research on the treatment of AD has remained a major focus area. As a marker of AD, Aβ was previously considered a key target for AD treatment, but the failure of clinical trials on such treatments has created doubts about the suitability of this target.172 For example, Roche has suspended most of its Aβ-related clinical trials (NCT05552157). However, Aβ-targeted strategies have not been abandoned completely; some clinical trials continue to focus on Aβ. For example, NCT05642429 was launched in 2022 to develop Aβ vaccine, and it is currently in phase I clinical trials. However, in response to the decline in attention to Aβ, research on other therapeutic targets, mainly neuroinflammation and p-Tau, has shown an upward trend. Furthermore, two studies targeting the gut microbiota (NCT03998423) progressed to phase IV, although NCT03998423 was discontinued because of the COVID-19 pandemic. The rapid development of biotechnology has also resulted in some clinical trials dedicated to the development of biological agents for AD theranostics. For example, NCT04040348 is exploring the role of human MSCs in the treatment of AD, and NCT04388982 seeks to develop MSC-derived exosomes with anti-AD efficacy. Because the preparation of these biological agents requires strict control of sterility, avoidance of external stimulation, and rare raw materials, this strategy is more expensive than the development of herbal medicines.
In addition, highly anticipated artificial nanocarriers have also been applied to AD theranostics. For example, one clinical trial is investigating a nano-drug delivery system of APH-1105 (NCT03806478). Irrespective of whether naturally derived exosomes or artificial nanomedicines are applied, their clinical transformation is the most important aspect of personalized AD diagnosis and treatment. Once the effectiveness of an agent is confirmed, AD theranostics will be revolutionized. However, it is worrisome that almost all anti-AD drugs under clinical investigation focus on only one or a few targets, and only one study has attempted to remodel the AD microenvironment by improving lesion metabolism (NCT05040321). Nonetheless, many clinical trials based on different drug dosage forms and targets have been conducted in the past 5 years (Table 3), indicating that AD therapy is gradually moving toward multi-target and diversification approaches.
| Clinical trial identifier | First posted | Agent | Intervention target | Phase | Status |
|---|---|---|---|---|---|
| NCT05728736 | 2023 | Lemborexant | N/A | Phase II | Recruiting |
| NCT05469360 | 2022 | NIO752 | N/A | Phase I | Recruiting |
| NCT03748706 | 2018 | PTI-125 | N/A | Phase II | Completed |
| NCT03461276 | 2018 | ABvac40 | Aβ | Phase II | Unknown |
| NCT03518073 | 2018 | Zagotenemab | N/A | Phase II | Completed |
| NCT03444870 | 2018 | Gantenerumab | N/A | Phase III | Completed |
| NCT03493282 | 2018 | CT1812 | Regeneration | Phase I/II | Completed |
| NCT03435861 | 2018 | VX-745 | Neuroinflammation | Phase II | Unknown |
| NCT03806478 | 2019 | APH-1105 nanoparticles | N/A | Phase II | Not yet recruiting |
| NCT04098666 | 2019 | Extended-release metformin | N/A | Phase II/III | Recruiting |
| NCT03991988 | 2019 | Montelukast | N/A | Phase II | Completed |
| NCT04191486 | 2019 | T-817MA | p-Tau | Phase II | Active, not recruiting |
| NCT04079803 | 2019 | Simufilam | N/A | Phase II | Completed |
| NCT03856359 | 2019 | Rifaximin | Inflammation | Phase II | Completed |
| NCT04040348 | 2019 | Approximately 100 million allogeneic human mesenchymal stem cells (MSCs) | N/A | Phase I | Active, not recruiting |
| NCT03790709 | 2019 | ANAVEX2-73 | N/A | Phase II/III | Completed |
| NCT03998423 | 2019 | Fecal microbiota transplant | Gut microbiota | Phase I | Terminated |
| NCT04390646 | 2020 | Gonadotropin-releasing hormone, gonadorelin acetate | Regeneration | Phase II | Active, not recruiting |
| NCT04517552 | 2020 | [11C]-CS1P1 | Neuroinflammation | N/A | Recruiting |
| NCT04619420 | 2020 | JNJ-63733657 | p-Tau | Phase II | Recruiting |
| NCT04685590 | 2020 | Dasatinib + Quercetin | N/A | Phase II | Recruiting |
| NCT04437511 | 2020 | Donanemab | p-Tau | Phase III | Active, not recruiting |
| NCT04468659 | 2020 | Lecanemab | N/A | Phase III | Recruiting |
| NCT04629547 | 2020 | Suvorexant | Aβ | Phase II | Recruiting |
| NCT04570644 | 2020 | ALZT-OP1 (cromolyn and ibuprofen) ALZT-OP1a (cromolyn) and ALZT-OP1b (ibuprofen) | N/A | Phase I | Completed |
| NCT04413344 | 2020 | Bromocriptine | Aβ | Phase I/II | Completed |
| NCT04308304 | 2020 | MK-1942, donepezil | N/A | Phase I | Completed |
| NCT04500847 | 2020 | Emtriva capsule | Neuroinflammation | Phase I | Recruiting |
| NCT04381468 | 2020 | Pepinemab | N/A | Phase I/II | Recruiting |
| NCT04251182 | 2020 | T3D-959 | N/A | Phase II | Completed |
| NCT04388982 | 2020 | MSC-derived exosomes | N/A | Phase I/II | Unknown |
| NCT04592874 | 2020 | AL002 | N/A | Phase II | Recruiting |
| NCT05063539 | 2021 | LY3372689 | N/A | Phase II | Active, not recruiting |
| NCT05143528 | 2021 | Nilotinib BE | N/A | Phase III | Not yet recruiting |
| NCT04693520 | 2021 | ALZ-801 | N/A | Phase II | Active, not recruiting |
| NCT05040217 | 2021 | AAV2-BDNF | Regeneration | Phase I | Recruiting |
| NCT05108922 | 2021 | Donanemab, Aducanumab | Aβ | Phase III | Active, not recruiting |
| NCT04786223 | 2021 | C-11 ER-176 | Neuroinflammation | Phase II | Enrolling by invitation |
| NCT05026177 | 2021 | Simufilam | N/A | Phase III | Recruiting |
| NCT04770220 | 2021 | ALZ-801 | N/A | Phase III | Active, not recruiting |
| NCT05074498 | 2021 | TB006 | N/A | Phase I/II | Completed |
| NCT04780399 | 2021 | Yangxue Qingnao pills | N/A | Phase II | Recruiting |
| NCT05040321 | 2021 | MIB-626 | Mitochondria, Aβ, neuroinflammation, regeneration, insulin | Phase I/II | Recruiting |
| NCT04973189 | 2021 | SHR-1707 | N/A | Phase I | Recruiting |
| NCT05081219 | 2021 | Humulin® R U-100, empagliflozin | Vessel | Phase II | Recruiting |
| NCT04994483 | 2021 | Simufilam | N/A | Phase III | Recruiting |
| NCT04711486 | 2021 | Contraloid acetate | N/A | Phase I | Completed |
| NCT05606341 | 2022 | CpG1018 | N/A | Phase I | Recruiting |
| NCT05310071 | 2022 | Aducanumab | N/A | Phase III | Recruiting |
| NCT05531656 | 2022 | CT1812 | N/A | Phase II | Not yet recruiting |
| NCT05256134 | 2022 | Gantenerumab | N/A | Phase III | Active, not recruiting |
| NCT05331144 | 2022 | Angiotensin II receptor blockers (ARBs, losartan) and calcium channel blockers (CCB, amlodipine) | Brain perfusion, Aβ, p-Tau | Phase II | Recruiting |
| NCT05291234 | 2022 | ABBV-916 | N/A | Phase II | Recruiting |
| NCT05194163 | 2022 | MW150 | p38 alpha MAPK kinase | Phase II | Not yet recruiting |
| NCT05423522 | 2022 | NanoLithium® NP03 | N/A | Phase II | Recruiting |
| NCT05189210 | 2022 | GV1001 | Aβ | Phase II | Recruiting |
| NCT05400330 | 2022 | LX1001 | Gene therapy | Phase I | Not yet recruiting |
| NCT05642429 | 2022 | AV-1959D | Aβ | Phase I | Recruiting |
| NCT05806177 | 2023 | AD16 | N/A | Phase I | Completed |
| NCT05681819 | 2023 | SHR-1707 | N/A | Phase I | Not yet recruiting |
- Note: Information obtained from the website ClinialTrials.gov.
10 CONCLUSION AND FUTURE PERSPECTIVES
Leveraging nanotechnology allows enhanced precision and detection limits in the design of nanosensors and nanoprobes, enabling the development of more accurate detection methods for pathological Aβ, NFTs, and other AD markers. These advances will enable earlier detection of AD, perhaps at a lower cost, facilitating early diagnosis and treatment and thus improving patient prognosis. More importantly, the application of nanotechnology has revolutionized the development of anti-AD drugs. The primary objective of employing nanotechnology in AD therapy is to develop delivery systems that can cross the BBB and enhance drug accumulation in the brain. Recent scientific advancements, have allowed researchers to focus on the distribution of nanomedicines within the brain parenchyma, aiming to prevent drug degradation in lysosomes and achieve higher drug potency. In addition to serving as drug carriers, some nanomaterials possess inherent anti-AD effects.
In this review, we classified these nanomedicines for AD treatment according to the pathogenic factors they modify. To clear pathogenic proteins, nanomedicines can be developed into nanochaperones that chelate pathogenic proteins and thus inhibit further misfolding. They can also be used to fabricate nanosponges that adsorb these proteins and promote their degradation. Moreover, nanomaterials have also been leveraged to develop innovative techniques, such as PDT and SDT. However, the safety and efficacy of these strategies, particularly ROS-dependent PDT and SDT, require further validation.
Nanomedicines also allow the precise delivery of anti-apoptotic and anti-inflammatory drugs, providing an excellent platform for combating neuronal apoptosis and neuroinflammation. In addition, nanozymes can alleviate the expression of downstream apoptotic or pyroptotic factors by scavenging ROS, thereby achieving drug-independent attenuation of neuronal apoptosis and neuroinflammation. However, neuroinflammation is not caused solely by microglia and astrocytes; peripheral immune responses also play a crucial role. Notably, nanomedicines offer a broad range of tools, such as nanovaccines, for modulating the immune function of lymphocytes. In addition to delivering nutritional factors and genetic drugs, nanomaterials exert biological effects, promoting tissue regeneration and remodeling. Although most regeneration studies have not focused on pathogenic factor clearance, nanoplatforms capable of promoting tissue regeneration and simultaneously removing pathogenic factors are important and warrant further development. Their development of such nanoplatforms is feasible because of the multifunctional nature of nanomedicines, which gives them functionality beyond targeted delivery.
- 1)
Safety of nanomedicines: Although most studies have evaluated the biocompatibility of anti-AD nanomedicines in animals, fully comprehending their clinical biofate and potential safety concerns is challenging because of the differences between animals and humans. Moreover, the surface potential, particle size, and roughness of nanomedicines can affect their biodistribution. Furthermore, nanomedicines (especially those based on inorganic nanomaterials) may generate metabolites that cause adverse reactions. Therefore, the balance between the benefits of nanomedicine-based therapy and their potential toxic effects warrants further exploration.
- 2)
Nanomedicine preparation: At present, achieving reproducibility in the preparation process of most anti-AD nanomedicines is challenging. Even when nanomedicines are prepared on a small scale in laboratory settings, the obtained nanomedicines are still heterogeneous.
- 3)
Drug availability: Loaded drugs are a major contributor to the efficacy of nanomedicines. In some cases, nanocarriers only act as drug delivery vehicles, and drugs with a higher potency can be more effective. Therefore, it remains necessary to explore and develop more suitable drug molecules for the treatment of AD.
- 4)
Targeting efficiency of nanoplatforms: Drugs should be able to target three key factors: lesions, diseased cells, and pathogenic factors. In AD, nanomedicines must cross the BBB to reach the lesion site. Therefore, anti-AD nanomedicines have lower targeting efficiency than nanomedicines for other diseases. Although several agents that simultaneously target the BBB, neurons, and microglia have been discovered, their specificity remains questionable. For instance, RVG29 can target neurons and microglia through their acetylcholine receptors, but it cannot distinguish diseased cells from healthy ones. Consequently, the drug may accumulate at higher levels in healthy cells. Notably, some special injection routes—such as nasal injection and intrathecal injection—can be used to bypass the BBB. When these injection strategies are adopted, modifications for crossing the BBB are not required; therefore, the nanomedicine design can only focus on targeting diseased cells, simplifying the formulation. However, apart from nasal delivery, these injection routes are invasive. Moreover, nasal delivery is also associated with several challenges, such as mechanisms of brain entry are unknown and suitable syringes may not be available. Hence, it is crucial to establish more specific targeting strategies.
- 5)
Systemic immune regulation: Most studies on neuroinflammation have focused solely on the brain's local immune environment. However, neuroinflammation not only exacerbates CNS disorders but also causes the release of chemokines that regulate and recruit peripheral immune cells. Although research has shown that peripheral lymphocytes, granulocytes, and macrophages infiltrate AD lesions, their role in AD progression remains poorly understood. Consequently, few studies have investigated the effect of these exogenous immune cells in alleviating neuroinflammation. Moreover, because the gut is the largest immune organ in the body, researchers have started exploring the role of the gut in neuroinflammation, and the brain–gut axis has become a major research hotspot. Nevertheless, it remains unclear how other immune organs, such as lymph nodes, the thymus, and the spleen, affect neuroinflammation. Moreover, no studies have explored whether these organs can be used as therapeutic targets for immune regulation. Thus, further studies are needed.
- 6)
Continued development of new anti-AD strategies: The distinctive physiological characteristics of the AD pathological microenvironment enable the development of corresponding therapeutic strategies. Although some characteristics, such as high ROS levels, are well-established, other aspects of the microenvironment have received less attention. Notably, compromised neurovascular units lead to an insufficient energy supply in the brain, resulting in local hypoxia and metabolic disturbances. Alleviating hypoxia and restoring metabolic processes may restore the energy levels required for vital activities, consequently reviving energy-dependent self-monitoring functions, such as the ubiquitin-proteasome system. Although restoring mitochondrial function can ameliorate metabolic dysfunction, it does not mitigate the issue of inadequate oxygen delivery. One potential solution involves the delivery of oxygen-carrying or oxygen-producing nanomaterials to the affected areas, allowing oxygen production in situ and alleviating metabolic disorders. Furthermore, a comprehensive understanding of AD could facilitate the development of more effective anti-AD strategies; this would provide more systemic and refined AD treatment approaches and improve their clinical efficacy.
- 7)
Effectiveness of pre-clinical models: The use of animal models is one of the most substantial challenges in AD research. Current modeling methods, which typically involve drug treatment or gene editing, cannot fully reproduce all the pathological features of AD. For example, 3xTg mice first develop Aβ and NFT lesions and subsequently show downstream effects, such as oxidative stress and neuroinflammation. However, the primary trigger of AD in clinical models and human patients is not clear. Hence, these modeling methods induce conditions that are different from clinical AD. Therefore, although animal experiments can yield positive outcomes, these positive outcomes may not necessarily translate to clinical efficacy. Hence, it is necessary to explore animal models that more closely resemble clinical AD.
In summary, nanotechnology offers novel avenues of therapy and more suitable strategies for AD treatment, as demonstrated in preclinical studies. Currently, although the number of studies is limited, clinical trials based on a variety of therapeutic targets and strategies are underway, providing hope for the gradual clinical translation of anti-AD nanomedicines. Thus, researchers must pay close attention to the latest evidence regarding AD. Nanotherapy is at the intersection of multiple disciplines and thus holds great potential for AD treatment. In the future, the clinical transformation of more precise and multi-targeted nanomedicines will help researchers address this significant neurodegenerative disorder and usher in a new era of AD management.
AUTHOR CONTRIBUTIONS
Guowang Cheng: Writing – original draft. Aihua Xie: Writing – original draft. Zhao Yan: Resources. Xiaozhen Zhu: Resources. Yafang Song: Resources. Tongkai Chen: Writing – review & editing.
ACKNOWLEDGMENTS
We gratefully acknowledge the support from the Guangdong Basic and Applied Basic Research Foundation (2019B1515120043), the Key Fields of the Biomedicine and Health Foundation of Colleges and Universities in Guangdong Province (2022ZDZX2017 and 2021ZDZX2032), and the Natural Science Foundation of Guangdong Province (2023A1515011127).
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no competing interests.
Open Research
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.

