

Stereocontrol in Conformationally Stable C(sp2)─C(sp3) Atropisomers
Graphical Abstract
Building in our expertize in the synthesis of trans-dihydrobenzofurans via organocatalyzed domino Michael/O-cyclization between β-naphthols and nitroalkenes, we developed an efficient strategy to access a new family of atropisomers bearing a conformationally stable C(sp2)─C(sp3) bond and two stereogenic centers through simple and highly diastereoselective alkylation of dihydrofurans with structurally diverse electrophiles. This crucial step places the nitro group in a relative cis-configuration with respect to the aromatic moiety, effectively increasing the barrier to diastereomerization and stabilizing the molecular conformation.

Abstract
We report a two-step sequence for the enantioselective construction of a rare family of stable C(sp2)─C(sp3) atropisomers featuring two additional stereogenic centers. The process begins with an organocatalyzed dihydrobenzofurannulation that establishes and controls the stereochemistry of two carbon centers, followed by a highly diastereoselective functionalization that significantly enhances the barrier to diastereomerization of the C(sp2)─C(sp3) bond, effectively locking and stabilizing its configuration.
Introduction
Atropisomerism, a form of stereoisomerism arising from restricted rotation around single bonds, plays a pivotal role in modern organic chemistry, particularly in pharmaceutical development,[1-4] catalyst and ligand design,[5-8] and functional materials.[9-14] While significant progress has been made in the enantioselective synthesis of C(sp2)─C(sp2) atropisomers,[15-17] the enantioselective construction of less common atropisomeric families,[18, 19] particularly C(sp2)─C(sp3) atropisomers, remains largely underexplored, despite their occurrence in various natural products.[20-24] Notably, the restricted rotation around the C(sp2)─C(sp3) bond in bisnicalaterines B[25-27] gives rise to a distinctive combination of central and axial stereogenic elements, both centered on a single atom (Scheme 1a).[28, 29] This remarkable stereochemical feature is also found in cyclodepsipeptide ustiloxins,[30] where atropisomerism naturally occurs exclusively in the syn form.[31] Another notable example is the streptorubin B, which exists as a 10:1 mixture of atropisomers with a barrier to diastereomerization of 86 kJ.mol−1.[32-34] The enantioselective access to molecules displaying a stereogenic C(sp2)─C(sp3) bond remains largely unexplored, with only a few examples reported in the literature. This scarcity is primarily attributed to the low barriers to diastereomerization associated with this peculiar families of atropisomers.[35-39] Both our group[40] and later Hayashi's team[41-43] have encountered these challenges, leveraging C(sp2)─C(sp3) atropisomers with low diastereomerization barriers as intermediates to control stereogenic axes in classical biaryl atropisomers. In 2017, Bencivenni reported the first example of thermodynamic control over the C(sp2)─C(sp3) stereogenic axis (Scheme 1b).[44]

They achieved this breakthrough via an enantioselective Friedel–Crafts-type alkylation of suitably functionalized inden-1-ones with 8-substituted 2-naphthols, employing a chiral primary amine derived from quinidine as the catalyst. Alternatively, Sparr developed an elegant enantioselective rhodium-catalyzed [2 + 2 + 2] cyclotrimerization, enabling the exclusive formation of a single stereoisomer among six possible isomers, with stereocontrol reaching an impressive 0:0:2:98:0:0 stereoisomeric ratio.[45] This discovery was achieved through the design of chiral 9-aryltripticene derivatives, featuring a C(sp2)─C(sp3) stereogenic axis stabilized by six high rotational barriers, thereby preventing interconversion between diastereomers. Finally, Jørgensen's group published one of the first examples of multistereogenic synthesis incorporating a conformationally stable C(sp2)─C(sp3) bond alongside two consecutive stereogenic centers.[46] Their organocatalytic strategy facilitated the addition of 5H-benzo[a]pyrrolizine-3-carbaldehydes to Michael acceptors, leading to the corresponding cyclized product with good yield (up to 69%), and moderate to excellent enantio- and diastereoselectivity (up to 99% ee, from 1:1 to >20:1 dr). Methodologies specifically tailored for the enantioselective synthesis of C(sp2)─C(sp3) atropisomers remain scarce, and achieving both high enantioselectivity and scalability continues to be a challenging task.[47] The design of reactions capable of generating structurally diverse C(sp2)─C(sp3) atropisomers with broad functional group compatibility is critical for expanding their synthetic utility. Based on our interest in the synthesis of trans-dihydrobenzofuran 3 via an organocatalyzed domino Michael/O-cyclization between naphthol 1 and chloronitrolakenes 2,[48-53] we design a streamlined strategy to construct a new family of molecules incorporating multiple stereogenic elements. This approach involves a simple diastereoselective alkylation of dihydrobenzofurans 3 with an electrophile, yielding compounds that contain a conformationally stable C(sp2)─C(sp3) bond and two stereogenic centers. Notably, this key transformation positions the nitro group in a cis-configuration relative to the aromatic ring, effectively increasing the barrier to diastereomerization and stabilizing the atropisomeric conformation.
Results and Discussion
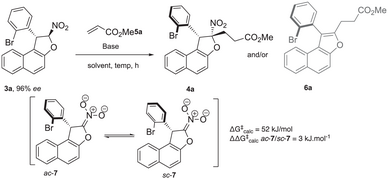
Based on the above considerations, preliminary computational studies were performed on dihydrobenzofurans 3a and 4a.[54] Dihydrobenzofurans 3a and 4a can adopt two conformations, anticlinal (ac) and synclinal (sc), resulting from rotation around the C─C bond (Scheme 2a). According to the Klyne–Prelog nomenclature,[55] the conformation is defined by the dihedral angle between the red and blue bonds. The major conformer, ac-3a, adopts an anticlinal conformation, whereas the minor conformer, sc-3a, exhibits a synclinal conformation. Notably, only the ac-3a diastereomer was detected by 1H NMR, probably due to the low barrier to diastereomerization calculated at 60.1 kJ.mol−1 and the significant energy difference between the ac-3a and sc-3a diastereomers (ΔΔG = 6.0 kJ.mol−1, Scheme 2b). Dihydrobenzofuran ac-4a was synthesized via a Michael addition between 3a and 5a under basic conditions. As expected, the relative cis configuration between the NO2 and 2-bromophenyl groups significantly increased the barrier to diastereomerization up to 163.0 kJ.mol−1 between the ac-4a and sc-4a diastereomers. Moreover, the potential energy of sc-4a was found to be 19.8 kJ.mol−1 higher than that of ac-4a (Scheme 2c), indicating that ac-4a is thermodynamically favored, with an abundance ratio of approximately 99.9:0.1 at 23 °C. In addition, as shown by the IGM maps (Scheme 2c),[56, 57] the relative cis configuration between NO2 and 2-bromophenyl groups greatly enhance the barrier to diastereomerization. The IGM map analyzes the gradient of the electron density in a molecular system to identify regions of space where chemical interactions take place. The IGM map is much larger and more repulsive, with even some strongly repulsive red areas, in 4a compared to that for 3a.

Building on these promising preliminary computations, we proceeded to optimize the Michael addition between the model substrates 3a and 5a as the Michael acceptor (Table 1).[58, 59] Under basic conditions, the chiral nitronate intermediate 7 is generated and subsequently trapped by the electrophilic acrylate 5a, which approaches from the opposite side of the 2-bromophenyl moiety. DFT calculations predicted the barrier to diastereomerization between nitronates ac- and sc-7 at 52 kJ.mol−1. As expected, this barrier is lower than the one of the substrate 3a because the removal of the proton by the base leads to less steric hindrance to go from one to the other conformer. We also found that ac- and sc-nitronates 7 have a small difference in energy, with a preference for the ac conformer, which corresponds to an ac/sc ratio of 4:1 at 298 K. When DBU was used as the base, exclusive formation of the benzofuran atropisomer 6a was observed (76% yield), likely resulting from the elimination of HNO2 from 4a through an E2-type mechanism (entry 1). In contrast, employing weaker bases selectively yielded the desired product 4a only when K2CO3 was used, affording a moderate yield (50%, entry 5) while maintaining high enantiopurity (96% ee) (entries 2–5). Switching to hydroxide bases led to better yields while minimizing degradation (entries 6 and 7). Further optimizations, including lowering the temperature to 0 °C (entry 8) and increasing the number of equivalents of 5a (3 equiv, entry 9), resulted in an enhanced yield.
 |
|||||
| Entry | Base | Equiv of base | Solvent | Yield of 4a (%)b) | ee of 4a (%)c) |
|---|---|---|---|---|---|
| 1d) | DBU | 1.2 | CH3CN | 0 | nd |
| 2 | tBuOK | 1.5 | DMF | <5% | nd |
| 3 | PPh3 | 0.4 | DMF/iPrOH (1/1) | 0 | nd |
| 4 | Et3N | 1.2 | THF/tBuOH (3/1) | 0 | nd |
| 5 | K2CO3 | 1.2 | THF/tBuOH (3/1) | 50 | 96 |
| 6 | Bu4NOH | 0.2 | THF/tBuOH (3/1) | 63 | 96 |
| 7 | Triton B | 0.2 | THF/tBuOH (3/1) | 68 | 96 |
| 8e) | Triton B | 0.2 | THF/tBuOH (3/1) | 68 | 96 |
| 9e), f) | Triton B | 0.2 | THF/tBuOH (3/1) | 76 | 96 |
| 10e), f) | Bu4NOH | 0.2 | THF/tBuOH (3/1) | 80 | 96 |
| 11e), f) | NaOH | 0.2 | THF/tBuOH (3/1) | 63 | 96 |
| 12e)-g) | Bu4NOH | 0.2 | THF | 74 | 96 |
| 13f)-h) | Bu4NOH | 0.2 | THF | 78 | 96 |
- a) Reaction conditions: 3a (1.0 equiv), 5a (1.5 equiv) and base were combined with solvent [0.1 M] at 20°C for 1 h.
- b) Isolated yields of analytically pure product after purification by flash chromatography.
- c) Determined by HPLC on chiral stationary phase.
- d) 76% of 6a was formed.
- e) reaction was run at 0°C.
- f) 3 equiv of 5a.
- g) 10% of 6a was obtained.
- h) reaction was run at −10°C.
At this temperature, tetrabutylammonium hydroxide outperformed Triton B (entry 10), whereas sodium hydroxide caused partial degradation and a reduced yield (entry 11). Notably, conducting the reaction in THF alone resulted in the formation of elimination product 6a in low yield (entry 12), even at lower temperatures (entry 13). Based on these results, the optimized conditions from entry 10 were selected to explore the reaction scope.
With the optimized reaction conditions in hand, we explored the generality of this enantioselective approach to novel C(sp2)─C(sp3) atropisomers with diverse nitroalkenes (Scheme 3). Other 2-substituted aryl groups containing chlorine and iodine atoms gave dihydrobenzofurans 4b and 4c with good yields and excellent stereoselectivity (>20:1 dr, >94% ee). Moroever, the incorporation of a nitro group in 4d and a methyl group in 4e resulted in comparable efficiency (>20:1 dr, >94% ee). In contrast, the incorporation of a 2-methoxynaphthyl group afforded a non-separable mixture of atropisomers sc-4g and ac-4g with a lower yield and diastereoselectivity (2:1 dr),[60] while maintaining high enantioselectivity (96% ee). In this case, the use of a 2-methoxy naphthyl group makes substrate 3g much less reactive, which could explain the lower yield. In addition, stereodifferentiation upon C─C bond formation during the Michael addition is less efficient as both ortho-positions are substituted, which could explain the low diastereoselectivity. Subsequently, the incorporation of a bromo substituent at either the 6-position or the 3-position of the naphthol ring furnished atropisomers 4h and 4i with moderate yields and excellent diastereoselectivity (>20:1 dr). However, the position of the bromo substituent had an impact on enantioselectivity, which slightly decreased from 96% (4h) to 80% (4i). Afterward, we explored the substrate scope with various Michael acceptors. The reaction proceeded efficiently with enone 5b and acrylonitrile 5c, affording functionalized atropisomers 4j and 4k, respectively, with comparable yields and high stereoselectivity (>20:1 dr, 95% ee). Extending the protocol to the Baylis–Hillman adduct 5d required modified conditions, employing K₂CO₃ in acetone at 23 °C. Under these conditions, the bromo- and iodo-derivatives 4l and 4m were obtained with good yields and excellent diastereo- and enantioselectivity (>20:1 dr, >94% ee). Next, an alternative C─halogen bond formation strategy was explored to further expand the synthesis of C(sp2)─C(sp3) atropisomers. Notably, applying the previous reaction conditions in the presence of N-chlorosuccinimide 5e enabled the formation of stable tertiary alkyl chlorides 4n and 4o with excellent yields (>87%) and high enantioselectivities (>92% ee), though in these two cases, a slight decrease in diastereoselectivity was observed. The halogenation was also possible using NBS, with the clean formation of 4p in 89% yield, slightly better diastereoselectivity ( 8:1 dr) and excellent enantioselectivity (96% ee). Additionally, the use of an aromatic aldehyde as an electrophilic partner proved feasible, introducing an additional stereocenter in 4q (1.7:1 dr, 97% ee), albeit with a moderate yield (27%). Finally, single-crystal X-ray diffraction analysis of atropisomers 4a, 4c, and 4l unambiguously confirmed their absolute configurations as ac,R,R.[61]

Finally, not only 2-naphthol derivatives could be incorporated in the final structure but also 1,3-dicarbnonyls, even if the later led to lower barriers to diastereomerization around the C(sp2)─C(sp3) bond (vide infra) and required the use of o,o’-disubstituted aryl groups on the starting nitroalkene 2 (Scheme 4). Remarkably, atropisomer sc-4r was obtained in 75% yield with high stereoselectivity (>20:1 dr, 84% ee) when 2-hydroxy-1,4-naphthoquinone was used as the starting material. Similarly, extending the protocol to dimedone led to the formation of atropisomer sc-4s with 64% yield. Although its diastereoselectivity slightly decreased to 15:1, it retained excellent enantioselectivity (96% ee). In the case of 4s, VT-1H NMR clearly showed that heating a 15:1 diastereomeric mixture of sc-4s at 50 °C for several hours resulted in the formation of its rotamer ac-4s with a thermodynamic equilibrium set at 2.1:1 ratio, corresponding to an energy difference of 2.1 kJ.mol−1 (Scheme 5). This allowed us to experimentally determine its barrier to diastereomerization at 107.7 kJ.mol−1, equivalent to a half-life of racemization of 9 days at 25 °C (see Supporting Information for details). In complement, DFT calculations predicted the barrier to diastereomerization for sc-4s at 114.2 kJ.mol−1, showing a very good agreement between experiment and model. Similar VT-1H-NMR experiments were conducted on compound 4t, which displayed a higher barrier to diastereomerization of 132.3 kJ.mol−1, corresponding to a racemization half-life of 1 day at 110 °C (see Supporting Information). Finally, ac-4u was obtained in good yield (76%), although in this case, the diastereoselectivity between ac and sc atropodiastereomers was lower (ac-4u/sc-4u = 3.5:1). This is probably due to less marked difference in term of steric hindrance between bromo and naphthyl substituents.


To evaluate the robustness of the method, a gram-scale experiment was conducted (Scheme 6a). Starting from one gram of substrate 3c under standard conditions, ac-4c was obtained in 76% yield without any loss of enantiopurity. Additionally, post-functionalizations of atropisomers 4a and 4c were performed to evaluate the practical utility of the current synthetic method (Scheme 6b).

As part of these studies, we explored the E2-elimination of HNO2, a transformation previously observed during the optimization of the Michael addition (see Table 1). After optimization (see Supporting Information), treatment of ac-4a with DBU as base in THF at room temperature for 16 h afforded the expected benzofuran atropisomer (aR)-6a in 95% yield and complete retention of axial chirality.[62] Its configuration has been assigned by single crystal X-ray diffraction analysis (SCXRD),[61] and interestingly, it is opposite to nitrobenzofuran (aS)-8, which was obtained through oxidative central-to-axial chirality conversion,[52] using MnO2 as the oxidant. Under these previous conditions, the fast conformational equilibrium around the C(sp2)─C(sp3) bond in 3a allowed the selective formation of (aS)-8, following the Curtin–Hammett principle. In contrast, the current transformation involves the aromatization by E2 elimination of HNO2 to give (aR)-6a and occurs exclusively from ac-4a whose conformation around the C(sp2)─C(sp3) bond is blocked.
In complement, reduction of the nitro group in ac-4c using zinc and concentrated hydrochloric acid yielded diastereoselectively N-hydroxylactam (ac,R,S)-9 in good yield while maintaining enantiopurity,[63, 64] with the process occurring via inversion of configuration at the stereogenic carbon atom bearing the nitro group as shown by single crystal X-ray diffraction analysis.[61] This might arise by epimerization of the transient hemiaminal intermediate 10 via ring-opening/ring-closing process. Alternatively, the Suzuki cross-coupling of 4c with 4-methoxyphenylboronic acid catalyzed by Pd(PPh3)4 afforded the desired product 11 with 44% yield, with complete retention of enantiopurity (92% ee) and no detectable diastereomerization under the applied thermal conditions (100 °C, 3 h). These outcomes highlight the exceptional configurational stability of this class of novel C(sp2)─C(sp3) atropisomers.
Finally, the chiroptical properties of (ac,R,R)-4a were evaluated through ECD and VCD spectral measurements and compared with theoretical calculations (Figure 1, see Supporting Information for details). The measured VCD spectrum (in green) exhibited a strong correlation with the calculated spectrum, demonstrating a satisfactory agreement (Figure 1, top). Most of the calculated vibrational bands align well with the experimental data, except for two bands at approximately 1570 and 1200 cm−1, which were not accurately modeled. These discrepancies are likely due to the challenges in correctly modeling the vibrational modes of NO2 moiety, as well as the possible involvement of weak intermolecular interactions. Regarding ECD, the calculated spectra for the predominant conformations of the molecule were nearly identical (Figure 1, bottom). This observation can be attributed to the fact that the molecular orbitals responsible for the electronic transitions associated with the measured bands are primarily located on the rigid part of the molecule, with minimal contribution from the methyl propanoate moiety, which exhibits greater conformational flexibility. Consequently, by integrating VCD, ECD, and molecular modeling (DFT and TD-DFT), the absolute configuration of (ac,R,R)-4a was unambiguously confirmed, corroborating the assignment previously determined by single-crystal X-ray diffraction analysis.

Conclusion
In summary, we have developed a highly diastereo- and enantioselective strategy for the synthesis of a new family of challenging C(sp2)─C(sp3) atropisomers incorporating two additional stereogenic centers. This approach relies on an enantioselective organocatalyzed dihydrobenzofurannulation of simple precursors, followed by a highly diastereoselective Michael addition that accommodates a wide range of electrophilic partners, enabling precise control over C(sp2)─C(sp3) stereogenicity. Importantly, barriers to diastereomerization were accessed both experimentally and by theoretical calculations, demonstrating excellent configurational stability in most cases. Furthermore, these C(sp2)─C(sp3) atropisomers can be easily transformed into their corresponding benzofuran atropisomers through a straightforward DBU-promoted elimination, highlighting the versatility and synthetic utility of this methodology.
Supporting Information
The authors have cited additional references within the Supporting Information.[65-69]
Acknowledgements
Financial support from the Agence Nationale pour la Recherche (ANR-21-CE07-0036), Aix-Marseille Université, the Centre National de la Recherche Scientifique (CNRS), Centrale Marseille, and the Young Scientists Fund of the National Natural Science Foundation of China (No. 22302176) is gratefully acknowledged. The authors thank Dr. N. Vanthuyne and M. Jean (https://fr-chimie.univ-amu.fr/chiropole/) for chiral HPLC analysis. J.-C.C. gratefully acknowledges the partial financial support from the Universidad Pedagógica y Tecnológica de Colombia for performing the research stay. The Centre de Calcul Intensif d'Aix-Marseille (mesocentre) is acknowledged for granting access to its high-performance computing resources (project b289).
Conflict of Interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the supporting information of this article.

