

Tertiary Amides as Directing Groups for Enantioselective C−H Amination using Ion-Paired Rhodium Complexes
Graphical Abstract
Direct amination of benzylic C−H bonds generates important stereocenters. Our approach, in which the chirality of a bimetallic rhodium complex is located on its associated cations, achieves this on tertiary amide-containing substrates to give amination of arylbutyric and arylvaleric acid-derived amides. We believe that the amide most likely interacts directly with the chiral cation to provide a highly organised transition state for C−H amination.

Abstract
Enantioselective C−H amination at a benzylic methylene is a vital disconnection towards chiral benzylamines. Here we disclose that butyric and valeric acid-derived tertiary amides can undergo highly enantioselective benzylic amination using an achiral anionic Rh complex that is ion-paired with a Cinchona alkaloid-derived chiral cation. A broad scope of compounds can be aminated encompassing numerous arene substitutions, amides, and two different chain lengths. Excellent tolerance of ortho substituents was observed, which has not been achieved before in asymmetric intermolecular C−H amination with Rh. We speculate that the tertiary amide group of the substrate engages in hydrogen bonding interactions directly with the chiral cation, enabling a high level of organisation at the transition state for C−H amination. This is in contrast with our previous work where a substrate bearing a hydrogen bond donor was required. Control experiments led to the discovery that methyl ethers also function as proficient directing groups under the optimised conditions, potentially also acting as hydrogen bond acceptors. This finding has the promise to dramatically expand the applicability of our ion-paired chiral catalysts.
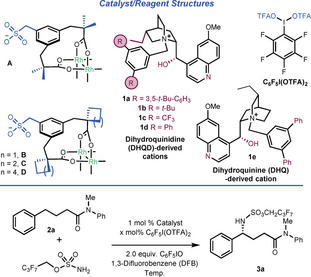
The direct introduction of amine functionality into simple building blocks produces compounds of importance in a range of areas.1 Direct C−H amination at benzylic positions is of particular interest—the benzylamine motif is prominent in numerous important compounds and a new stereocentre is normally produced. One of the leading methods for enacting C−H amination in an enantioselective manner is to use metal nitrenoids.2 A variety of metals have been explored for this purpose including ruthenium,3 silver,4 iridium,5 cobalt,6 manganese,7 and iron.3f, 8, 9 In recent years, enzymes have also been productively harnessed to enable asymmetric C−H amination.10 Within metals, Rh complexes have played a particularly important role in enabling practical intermolecular C−H amination reactions.11 Significant efforts have been made to develop enantioselective variants, most of which use chiral carboxylic acids to ligate a “paddlewheel” Rh tetracarboxylate dimer (Scheme 1a, left).3a, 12, 13 Distinct variations of this exploit outer-sphere interactions, including the use of a dual hydrogen bonded template by Bach14 and most recently a peptide-derived ligand for Rh by Miller.15 All of these approaches, despite their individual strengths, possess limitations and further development of asymmetric approaches is certainly warranted given the importance of chiral benzylamines.

Background and introduction.
We recently developed a conceptually distinct approach to enantioselective C−H amination in which Rh is ligated by two anionic, achiral bis-carboxylate ligands closely derived from Du Bois’ esp,16 the state-of-the-art ligand for intermolecular non-enantioselective amination.17, 18 This bisanionic complex is paired with chiral cations derived from quaternised Cinchona alkaloids, motifs proven as highly effective inducers of asymmetry in organocatalysis19 but which have rarely been explored in combination with transition metal catalysis (Scheme 1a, right).20, 21 Of the substrates tested in our original study, those that gave high enantioselectivity all featured a primary alcohol group and we speculated that the alcohol may act as a hydrogen bond donor to interact with the sulfonate group on the ligand. This would provide organisation at the C−H activation transition state located within the chiral pocket of the cation (Scheme 1b, left).22, 23 We also reported an example with a carboxylic acid as directing group which gave 78 % ee, albeit in very low yield (Scheme 1b, right). The precise nature of the interaction with the catalyst was not clear, but interaction of the carboxylic acid with the sulfonate group seemed plausible.
The finding that spurred the work described herein was the subsequent discovery that a control substrate bearing a methyl ester, now unable to act as a hydrogen bond donor, still gave a considerable 51 % ee in the benzylic amination under conditions from the previous study, albeit in very low yield (Scheme 1c). This observation raised the intriguing question as to whether, with further optimisation and development, we may be able to diversify the scope of the process away from alcohol-containing substrates. Our confidence was increased by our parallel findings in chiral cation directed C−H borylation, where promising enantioselectivity could be obtained in the borylation of a phosphine oxide substrate able to function only as a hydrogen bond acceptor.24 We speculated whether carbonyl functionality may engage with the ion-paired catalyst by direct interaction with the chiral cation through hydrogen bonding with the quinine hydroxyl, as well as potentially the α-CH bonds adjacent to the quaternised nitrogen (Scheme 1d, left). In turn, the chiral cation would remain ion-paired with the anionic Rh complex, maintaining a high level of organisation. If achievable, C−H amination of butyric acid-derivatives would lead directly to enantioenriched derivatives of γ-amino butyric acids, important motifs in medicinal chemistry.25 Here we demonstrate that, through modification of the conditions and catalyst, tertiary amides are highly proficient at directing asymmetric C−H amination. The reaction has remarkable functional group tolerance at all positions of the arene and is effective for two chain lengths (Scheme 1d, right). A series of control experiments led to the discovery that ethers are also viable directing groups under the present conditions.
We started by evaluating the amination of amide-containing substrates on the basis that an amide should be an excellent hydrogen bond acceptor. Accordingly, tertiary amide 2 a, derived from phenylbutyric acid, was tested in combination with ion-paired catalyst Rh2(D)2 ⋅ (1 a)2 which had been optimal in our previous alcohol-directed C−H amination (Table 1).17a While an encouraging yield of product 3 a was obtained at 0 °C in 1,3-difluorobenzene (1,3-DFB) as solvent, enantioselectivity was low (Table 1, entry 1). In recent studies on enantioselective aziridination of alkenyl alcohols using similar complexes we observed a beneficial effect of adding a sub-stoichiometric amount of a weak acid additive.17b C6F5I(OTFA)2 was found to be a convenient source of trifluoroacetic acid, releasing it as the oxidant is consumed.26 We were pleased to find that when using 20 mol % of C6F5I(OTFA)2 as an additive the ee was boosted substantially to 70 %, with the yield improved also (entry 2).27 Lowering the temperature to −25 °C gave further improvement (entry 3) and at this temperature we evaluated several other cations (1 b–1 d, entries 4–6), finding that the slightly smaller 1 d was optimal (entry 6, 84 % ee). Lowering the temperature further to −35 °C gave a marginal improvement but still with good reactivity (entry 7). Finally, an evaluation of dimer scaffolds A–C revealed that all were very similar (and slightly better than D), with cyclobutane-containing dimer B giving the optimal metrics (entries 8–10). The optimal cation 1 d was derived from dihydroquinidine, and we found that the same complex paired with the cation derived from pseudoenantiomeric dihydroquinine (1 e) gave the opposite product enantiomer upon amination, as expected, but with markedly lower enantioselectivity (entry 11, −77 % ee). We then reevaluated the optimised reaction in the absence of the 20 mol % of C6F5I(OTFA)2 additive and found that enantioselectivity was significantly reduced, confirming its important role (entry 12, 54 % ee). Substituting 20 mol % of C6F5I(OTFA)2 with 40 mol % of TFA gave a very similar outcome but we found that practically it was easier to weigh out small quantities of the former (entry 13).28 The ester substrate shown in Scheme 1c was reevaluated under the optimised conditions giving 24 % yield and 60 % ee, demonstrating the relative proficiency of the amide as a directing group.
|
|||||
Entry |
Temp (°C) |
X (mol %) |
Catalyst |
Yield (%)[a] |
ee (%)[b] |
|---|---|---|---|---|---|
1 |
0 |
0 |
Rh2(D)2 ⋅ (1 a)2 |
49 |
15 |
2 |
0 |
20 |
Rh2(D)2 ⋅ (1 a)2 |
95 |
70 |
3 |
−25 |
20 |
Rh2(D)2 ⋅ (1 a)2 |
97 |
77 |
4 |
−25 |
20 |
Rh2(D)2 ⋅ (1 b)2 |
90 |
70 |
5 |
−25 |
20 |
Rh2(D)2 ⋅ (1 c)2 |
90 |
72 |
6 |
−25 |
20 |
Rh2(D)2 ⋅ (1 d)2 |
96 |
84 |
7 |
−35 |
20 |
Rh2(D)2 ⋅ (1 d)2 |
97 |
85 |
8 |
−35 |
20 |
Rh2(A)2 ⋅ (1 d)2 |
98 |
90 |
9 |
−35 |
20 |
Rh2(C)2 ⋅ (1 d)2 |
98 |
91 |
10 |
−35 |
20 |
Rh2(B)2 ⋅ (1 d)2 |
99 (91) |
91 |
11 |
−35 |
20 |
Rh2(B)2 ⋅ (1 e)2 |
91 |
−77 |
12 |
−35 |
0 |
Rh2(B)2 ⋅ (1 d)2 |
57 |
54 |
13[c] |
−35 |
0 |
Rh2(B)2 ⋅ (1 d)2 |
100 |
88 |
- [a] Yields determined by 1H NMR using 1,3,5-trimethoxybenzene (TMB) as an internal standard. Value in parentheses corresponds to the isolated sample. [b] ee determined by chiral SFC analysis of the crude reaction mixture. [c] Trifluoroacetic acid (40 mol %) added.
The scope of the reaction was next explored, first examining tolerance with respect to N-substituents on the amide (Scheme 2A). This was extremely broad suggesting that the substituents may be pointing away from the bulk of the catalyst and into open space. A secondary amide gave poor yield but still reasonable enantioselectivity (3 b). Very bulky alkyl groups such as isopropyl (3 c) and cyclohexyl (3 d) could be incorporated without significant impact on ee. The presence of either two aryl (3 e) or alkyl (3 f) groups on the amide was tolerated and the latter could both be as bulky as isopropyls with only minimal reduction in ee (3 g). Cycloalkyls (3 h, 3 i) and a benzyl group (3 j) could also be included and a Weinreb amide-bearing substrate (3 k) performed particularly well, an excellent outcome given the versatility of this motif in synthesis.29

Scope studies on aryl butyric acid-derived amides. Encompassing: A) amide substitution, B) arene substitution and C) Weinreb amides.
Using 3 a as a basis we evaluated the scope of arene substitution (Scheme 2B). Excellent results were obtained and substituents with a wide variety of steric and electronic properties were tolerated in the para (4 a–4 i), meta (4 j–4 p) and ortho (4 q–4 v) positions, as well as some alternative aromatic (4 w) and heteroaromatic (4 x) systems. We were pleasantly surprised by the fact that substrates bearing strong resonance withdrawing groups such as ester (4 e), sulfone (4 i) and even nitro (4 h) delivered the products with excellent yields and ee, setting this apart from other leading Rh-catalysed C−H amination processes.12d, 12f, 12g, 15 Emphasising functional group compatibility, a BPin group could be incorporated with moderate yield (4 p). A wide range of substituents were tolerated at the ortho position without significant loss of reactivity, even with bulky groups such as phenyl (4 u) and isopropyl (4 v), and in fact enantioselectivity appeared to be boosted to the very highest levels (98 % ee in several cases). Again, this contrasts with other Rh-catalysed enantioselective benzylic amination protocols in which ortho substituents cannot be accommodated.12f, 15 We also demonstrate the effectiveness of the amination on a smaller selection of variously substituted Weinreb amide substrates, and here it was only with a large ortho-phenyl substituent (5 k) that yield and ee were impaired (Scheme 2C).
Our previous alcohol-directed C−H amination only delivered high enantioselectivities for a single chain length (four methylenes) between the arene and the alcohol—longer or shorter gave poorer outcomes.17a After a re-evaluation of quaternising groups on the cation (see SI) we found that complex Rh2(B)2 ⋅ (1 c)2 delivered very good selectivity for longer chain substrates derived from arylvaleric acids (Scheme 3). We explored examples featuring meta (7 b–7 d) and ortho (7 e–7 h) substitution, a naphthyl (7 i) and trisubstituted 7 j. The reaction was also effective on longer chain Weinreb amide-derived amides (7 k–7 m). Addition of an extra methylene into the chain of substrate 6 a reduced ee to 76 % (see SI).

Scope studies on arylvaleric acid-derived amides.
The reaction products are amenable to elaboration to give useful motifs (Scheme 4A). N-Deprotection could be achieved by simply heating in aqueous dioxane (8 a, 8 b).15 Heating these with catalytic acid forms the corresponding lactams (8 c and 8 d), from which the absolute stereochemistry could be assigned. Ditosylation of the amine (8 e) and heating in DMF promoted hydrolytic cyclisation with inversion of stereochemistry (8 f). More reactive reagents achieved reduction to the amine (LiAlH4) and Grignard addition to a Weinreb amide (Scheme 4B). Enantiopurity was retained throughout, showcasing the versatility of the enantioenriched building blocks obtained. As disclosed earlier, the pseudoenantiomeric dihydroquinine-derived cation 1 e gave lower ee of the antipode of 3 a (77 % vs. 91 %). We remedied this using the same approach that we took previously, inspired by prior work of Deng, by synthesising 1 f, the cation derived from desvinylquinine (Scheme 4C).30 The complex incorporating this cation gave the opposite enantiomer in 92 % ee.

Post-functionalisation of reaction products and access to product antipode.
Experiments were next carried out to compare amide- and alcohol-directed C−H amination using the presently optimised conditions, which include 20 mol % C6F5I(OTFA)2 additive. An intermolecular competition experiment between amide (2 a) and alcohol (9) substrates using limiting aminating agent showed little preference for one over the other, with both products obtained in high ee (Scheme 5A). An intramolecular version of this competition was then carried out using substrate 10 and gave a similar outcome, providing further indication that the ‘directing effect’ of the amide and alcohol appeared similar under these conditions (Scheme 5B).31 We then prepared a version of the optimal cation in which the hydroxyl group is methylated (1 g), precluding it from acting as a hydrogen bond donor (Scheme 5C, inset). Using the complex incorporating 1 g we reevaluated intramolecular competition substrate 10, probing whether this may disfavor amination on the amide-containing chain by precluding a possible hydrogen bonding interaction between amide and catalyst, but still allow amination on the alcohol-containing chain if the latter is acting as a hydrogen bond donor to the catalyst sulfonate group. Again, there was no site-selectivity preference, and interestingly both products were obtained in low ee (Scheme 5C). This poor enantioselectivity outcome when using the O-methylated cation was confirmed in separate experiments using the two individual substrates (see SI). These consistent observations between the two functional groups, combined with the fact that the same product absolute stereochemistry is obtained in both reactions (when using the same enantiomer of catalyst), hinted at the two groups potentially interacting with the catalyst in a similar manner under the present conditions. We therefore considered the possibility that the oxygen of the alcohol may function as a hydrogen bond acceptor, analogously to the amide, and so evaluated an O-methylated version of the intramolecular competition substrate with the optimal catalyst to test this (Scheme 5D, 11).32 This again gave both products and a very high 79 % ee was obtained for amination on the ether chain. In contrast, silylated substrate 12 gave amination selectively on the amide chain, possibly due to the bulky TBS group precluding hydrogen bonding interactions with the ether oxygen (Scheme 5E, upper). Use of Rh2(esp)2 on 12 gave very little selectivity between the two chains, a presumed result of an undirected reaction (Scheme 5E, lower). The ability of the methyl ether to direct the reaction under these conditions was confirmed in a separate experiment on simple O-methylated alcohol 13 which gave 85 % ee (Scheme 5F, upper), an almost identical ee outcome to that obtained under the same conditions with alcohol 9 (middle). Complete removal of the functional group in amination of ethylbenzene (14) gave very poor outcome in terms of both yield and ee (lower), emphasising that the ether is playing a directing role. Very interestingly, amination of 13 under the conditions used in our previous report on alcohol-directed amination (−25 °C, PhIO as oxidant, no C6F5I(OTFA)2 additive) gave very poor yield and ee, pointing to a key role played by the C6F5I(OTFA)2 additive in enabling the ether to be effective. We have already shown that this additive was crucial for high ee in the amide-directed reaction (Table 1, entries 1 and 12) and this appears necessary to allow hydrogen bond acceptor groups to be effective; in its absence, the hydrogen bond donor of the hydroxyl is required for high ee. To confirm this, we revisited the intermolecular competition experiment under our previous conditions, which did not include the C6F5I(OTFA)2 additive, and saw 8 : 1 selectivity in preference for the alcohol substrate, amination of which occurred in 74 % ee (Scheme 5G). The small amount of the amide amination that occurred did so with only 15 % ee, further reinforcing that the amide is rendered an effective director only in the presence of the weak acid additive. The precise origins of this are presently unclear but it is feasible that the presence of the acid significantly changes the chiral pocket produced by the cation and we are working to explore this further.

Experiments to probe the interactions crucial for high ee.
In summary, we demonstrate that tertiary amides are highly effective at engaging with chiral, ion-paired rhodium catalysts to enable highly enantioselective benzylic amination of a broad variety of arylbutyric acid and arylvaleric acid-derived amides. Direct comparison of alcohol- and amide-directed amination revealed similarities, and control experiments revealed that under the present optimised conditions an ether was also a proficient directing group.33 For hydrogen bond acceptor groups to direct with high ee, use of a weak acid-releasing additive was crucial; in its absence only a hydrogen bond donor alcohol is effective. We anticipate that this realisation that substrates bearing hydrogen bond acceptor groups can engage in a highly productive manner with the ion-paired catalyst significantly expands the applicability of these catalysts for asymmetric C−H amination and beyond.
Acknowledgments
We are grateful to Harry Palmer for the synthesis of several substrates that were used in this study, and to Dr. Alexander Fanourakis for invaluable discussions throughout. We are grateful to the European Research Council under the Horizon 2020 Program (Starting Grant no. 757381) for funding for this work. B.D.W. is grateful to the Cambridge Trust and Sidney Sussex College Cambridge for a PhD studentship.
Conflict of interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.

