

Photo-Organocatalytic Enantioselective Radical Cascade Reactions of Unactivated Olefins
Graphical Abstract
A radical approach: A photochemical asymmetric radical cascade process that converts unactivated olefins and α,β-unsaturated aldehydes into chiral adducts in a single step is reported. This enantioselective strategy, which is triggered by the excited-state reactivity of chiral iminium ion intermediates, combines two sequential radical-based bond-forming events.

Abstract
Radical cascade processes are invaluable for their ability to rapidly construct complex chiral molecules from simple substrates. However, implementing catalytic asymmetric variants is difficult. Reported herein is a visible-light-mediated organocatalytic strategy that exploits the excited-state reactivity of chiral iminium ions to trigger radical cascade reactions with high enantioselectivity. By combining two sequential radical-based bond-forming events, the method converts unactivated olefins and α,β-unsaturated aldehydes into chiral adducts in a single step. The implementation of an asymmetric three-component radical cascade further demonstrates the complexity-generating power of this photochemical strategy.
Cascade processes are powerful strategies for rapidly increasing structural and stereochemical complexity while delivering complex chiral molecules in one step.1 The high reactivity and selectivity of open-shell intermediates2 makes radical chemistry perfectly suited for implementing cascades.3 However, the intrinsic challenge of carrying out reactions with radicals in a stereocontrolled fashion4 has greatly limited the development of enantioselective variants. The majority of the reported strategies rely on either chiral substrates or stoichiometric ligands.3c, 4, 5 Recently, photoredox catalysis6 has provided effective tools to enable single-electron transfer (SET) mediated radical cascade processes under mild reaction conditions.3a The combination with chiral Lewis acid catalysts has allowed the ensuing radical cascade process to be channeled towards a stereocontrolled pattern.7 Although effective, these catalytic asymmetric methods are generally limited to either formal [2+2]7a or [3+2]7b,7c photocycloaddition reactions or cation radical Diels–Alder processes.7d, 8
Recently, our laboratories identified a new photochemical catalytic mode of substrate activation that exploits the excited-state reactivity of chiral iminium ions (I) to generate radicals upon SET oxidation of suitable substrates (II; Figure 1 a).9 The resulting chiral intermediate III can also govern the ensuing radical coupling step with high stereoselectivity. We wondered if this new photochemical activation mode could be used to expand the synthetic potential of enantioselective radical cascades beyond the reactivity constraints of formal cycloaddition chemistry.8 In an initial attempt, we combined the excited-state iminium ion chemistry with the ground-state reactivity of enamines to access cyclopentanols, from cyclopropanols, with high stereoselectivity (Figure 1 b).10 However, while the first carbon–carbon bond-forming step in this cascade sequence is a radical process, the subsequent carbon–carbon bond formation follows a two-electron pathway. Herein, we detail how the photochemistry of iminium ions has served to successfully implement an enantioselective catalytic radical cascade reaction that combines two sequential radical-based bond-forming processes without mechanistically relying on a cycloaddition manifold (Figure 1 c). This organocatalytic photochemical process converts readily available enals and unactivated olefins, bearing an oxygen-centered nucleophilic handle, into complex chiral molecules containing butyrolactone11a or tetrahydrofuran moieties.11b These two structural elements are present in numerous biologically active and naturally occurring molecules.

a) Our previous study demonstrated that light excitation turns iminium ions (I) into chiral oxidants, and the resulting SET-based mechanism for generating radicals from precursors II bearing a redox auxiliary (RA). b) A cascade reaction proceeding by an excited iminium ion/ground-state enamine sequence, where a stereocontrolled radical path is combined with polar reactivity. c) Proposed enantioselective catalytic radical cascade proceeding by two sequential radical steps: upon SET oxidation of an unactivated olefin, an anti-Markovnikov addition to the radical cation IV is followed by a stereocontrolled radical coupling. SET=single-electron transfer.
Figure 2 details our proposed strategy to develop an enantioselective SET-triggered radical cascade. We envisioned a catalytic cycle wherein a chiral amine catalyst would form the colored iminium ion intermediate I upon condensation with the enal 1. Selective excitation with a violet-light-emitting diode (LED) turns I into a strong oxidant (I*), as implied by its reduction potential, which was estimated at about +2.4 V (E*red (I*/I.−) vs. Ag/Ag+ in CH3CN) using electrochemical and spectroscopic methods.9a Therefore, the photoexcited iminium ion can oxidize an alkene substrate (2)12 adorned with a suitable oxygen-centred nucleophilic handle (alkenes 2 used in this study have oxidation potentials ranging from +2.03 to +2.19 V vs. Ag/AgCl in MeCN, see section D in the Supporting Information for details). The SET activation of 2 would concomitantly form the chiral 5π-intermediate III and the alkene radical cation IV. The nucleophilic moiety within IV would then trigger a polar-radical-crossover cycloaddition,13, 14 capitalizing on the simultaneous polar and radical nature of such radical cation species. This process would proceed with an anti-Markovnikov selectivity to afford the more stable tertiary radical intermediate V.15 At this juncture, a stereocontrolled radical coupling with III would afford the cascade product 3 with high enantioselectivity. This mechanistic plan finds support in the high reactivity of the distonic intermediate IV, which has been extensively investigated by Nicewicz to develop anti-Markovnikov hydrofunctionalizations of olefins,14, 15, 16 and its tendency to initiate cycloaddition sequences leading to heterocyclic products.14, 16

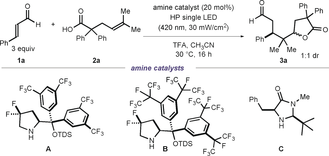
Mechanistic proposal for a SET-triggered asymmetric photocatalytic radical cascade process.
To test the feasibility of our photochemical plan, we selected cinnamaldehyde (1 a) and the gem-difluorinated diarylprolinol silylether catalyst A9a for the formation of the chiral iminium ion (Table 1). The experiments were conducted in CH3CN using a single high-power (HP) LED (λmax=420 nm) with an irradiance at 30 mW cm−2, as controlled by an external power supply (full details of the illumination set-up are reported in Figure S1 of the Supporting Information). The alkenoic acid 2 a was selected as the model substrate based on its oxidation potential (Epox (2 a.+/2 a)=+2.09 V vs. Ag/AgCl in MeCN) and its tendency to undergo SET oxidation mediated anti-Markovnikov lactonization.16a Using 40 % of trifluoroacetic acid (TFA) to assist the formation of I, the desired product 3 a was obtained at ambient temperature in 55 % yield, 88 % ee, and 1:1 d.r. (entry 1). Increasing the amount of TFA to 60 mol % improved the yield to 62 % (entry 2). The chiral amine catalyst B, possessing bulkier perfluoro-isopropyl groups on the arene scaffold, improved the enantioselectivity (91 % ee, entry 3), whereas the chiral imidazolidinone catalyst C afforded the product with low stereocontrol (entry 4). Implementation of a two-phase solvent system, using tetradecafluorohexane in a 1:1 ratio to CH3CN, further increased the yield of the reaction (72 % yield, entry 5).17 Control experiments indicated that both the light and the amine catalyst are required for reactivity (entries 7 and 8), while the addition of a radical trapping agent (TEMPO, 1 equiv) completely inhibited the process.
Entry |
Catalyst |
Reaction conditions |
Yield [%][b] |
ee [%][c] |
|---|---|---|---|---|
1 |
A |
40 % TFA |
55 |
88 |
2 |
A |
60 % TFA |
62 |
88 |
3 |
B |
60 % TFA |
63 |
91 |
4 |
C |
60 % TFA |
60 |
33 |
5 |
B |
CH3CN/HexF (1:1) |
72 (58) |
91 |
7 |
A |
no light |
0 |
– |
8 |
– |
no catalyst |
0 |
– |
- [a] Reactions performed on a 0.1 mmol scale at 30 °C for 16 h using 0.2 mL of solvent under illumination by a single high-power (HP) LED (λmax=420 nm) with an irradiance of 30 mW cm−2. [b] Yield of 3 a determined by 1H NMR analysis of the crude reaction mixture using trichloroethylene as the internal standard. Yields of the isolated 3 a are reported within parentheses. [c] Enantiomeric excess determined by HPLC analysis on a chiral stationary phase. HexF=tetradecafluorohexane, TDS=tert-hexyldimethylsilyl, TFA=trifluoroacetic acid.
Using the optimized reaction conditions described in entry 5 of Table 1, we then turned our attention to the scope of the photocatalytic asymmetric radical cascade process (Figure 3). We first evaluated the possibility of using differently substituted olefins. The presence of geminal methyl groups at the α-position of 2 significantly improved the yield and enantioselectivity (adduct 3 b, 75 % yield and 94 % ee). An ethyl fragment could also be installed (3 c), while the inclusion of a cyclohexyl group afforded the spirocyclic adduct 3 d in moderate yield and high enantioselectivity. The absence of α-substituents in 2 did not significantly affect the overall efficiency (product 3 e). This observation indicates that the Thorpe–Ingold effect is not critical to the cascade process. Concerning the substitution of the double bond, introducing a cyclohexyl in place of the dimethyl moiety slightly increased the diastereoselectivity to 1.3:1 (3 f) while maintaining a high chemical yield and enantioselectivity. Importantly, tetrahydrofuran derivatives 4 a and 4 b could also be synthesized when decorating the alkene substrate with an alcohol acting as the nucleophilic moiety.

Survey of the alkenoic acids and alkenols (2) that can participate in the radical cascade process with 1 a. Reactions performed on a 0.1 mmol scale using a 1:1 mixture of acetonitrile and tetradecafluorohexane. Yields and enantiomeric excesses of the isolated products 3 are indicated below each entry (average of two runs per substrate). Enantiomeric excesses measured on the enoate derivatives of 3, obtained upon olefination of the aldehyde with PPh3CHCO2Et. The d.r. value was inferred by 1H NMR analysis of the crude reaction mixture. [a] Reaction performed using the catalyst A in acetonitrile.
Crystals from the compound 3 a were suitable for X-ray crystallographic analysis,18 which established the stereochemical course of the radical cascade process.
Regarding the scope of the α,β-unsaturated aldehydes 1, moderately electron-withdrawing and electron-donating groups on the aromatic ring are tolerated without diminishing either the yield or enantioselectivity (Figure 4). The substituent can also be moved around the ring without impeding reactivity (3 g–l). Notably, synthetically useful groups, which can undergo further functionalization, were tolerated. These included halogens (3 g–j), trimethylsilyl (3 m), and pinacolborane (3 n). One limitation of this reaction is that aliphatic enals, including tert-butylacroleine and (E)-2-octenal, remained unreacted under the reaction conditions.

Survey of the enals 1 which can participate in the radical cascade process with 2 b. Reactions performed on a 0.1 mmol scale using a 1:1 mixture of acetonitrile and tetradecafluorohexane. Yields of the isolated products are indicated below each entry (average of two runs per substrate). Enantiomeric excesses measured on the enoate derivatives of adducts 3, obtained upon olefination of the aldehyde with PPh3CHCO2Et. The d.r. values were inferred by 1H NMR analysis of the crude reaction mixture.
We then focused on the design of a three-component radical cascade process to assemble complex chiral molecules from simple substrates in a single step. Specifically, we surmized that the excited iminium ion I* could oxidize amylene (5), an unfunctionalized alkene hydrocarbon (Epox (5.+/5)=+2.03 V vs. Ag/AgCl in MeCN; Figure 5).19 The anti-Markovnikov intermolecular addition of a carboxylic acid20 to the less substituted position of the resulting VI would then provide the key radical intermediate VII, which would eventually engage in a stereoselective radical coupling governed by the chiral 5π-intermediate III (structure shown in Figure 2). This catalytic asymmetric cascade process, which combines two intermolecular radical steps, would provide a one-step access to complex aldehydes (7).

Catalytic enantioselective three-component radical cascade process that combines two intermolecular radical steps. Reactions performed on a 0.1 mmol scale in CH3CN. Yields and enantiomeric excess given on the isolated enoate derivatives 8 obtained upon olefination of the aldehyde precursor (average of two experiments). The d.r. values were inferred by 1H NMR analysis of the crude reaction mixture. [a] Yield of the isolated intermediate aldehyde 7 b.
The use of α,α-difluorinated acids (6) enabled the successful realization of this enantioselective three-component cascade. The aldehydes 7 were converted into enoates 8 a–c, which could be isolated in moderate yields and enantioselectivities (Figure 5). The acids play a dual role in this transformation, since they assist the iminium ion formation while acting as nucleophilic substrates in the three-component cascade.
In summary, the photochemical activity of a chiral iminium ion has been exploited to activate unfunctionalized olefins and trigger an enantioselective radical cascade process. We expect that this photochemical organocatalytic activation mode could be useful for designing other radical cascade processes and for stereoselectively synthesizing more complex chiral scaffolds.
Acknowledgements
Financial support was provided by MINECO (CTQ2016-75520-P), and the European Research Council (ERC 681840—CATA-LUX). C.M.H. thanks the Marie Skłodowska-Curie Actions for a postdoctoral fellowship (H2020-MSCA-IF-2017 794211).
Conflict of interest
The authors declare no conflict of interest.

