

Catalytic 1,4-Rhodium(III) Migration Enables 1,3-Enynes to Function as One-Carbon Oxidative Annulation Partners in CH Functionalizations†
We thank the EPSRC for financial support (funding for D.J.B. through grant number EP/J018090/1, and a Leadership Fellowship to H.W.L.) and Martin D. Wieczysty at the School of Chemistry, University of Edinburgh, for preliminary investigations and assistance in the preparation of substrates.
Graphical Abstract
When two become one: 1,3-Enynes containing allylic hydrogen atoms cis to the alkyne are shown to act as one-carbon partners, rather than two-carbon partners, in various rhodium-catalyzed oxidative annulations. The mechanism of these unexpected transformations is proposed to occur through double CH activation, involving a hitherto rare example of the 1,4-migration of a RhIII species.

Abstract
1,3-Enynes containing allylic hydrogens cis to the alkyne are shown to act as one-carbon partners, rather than two-carbon partners, in various rhodium-catalyzed oxidative annulations. The mechanism of these unexpected transformations is proposed to occur through double CH activation, involving a hitherto rare example of the 1,4-migration of a RhIII species. This phenomenon is general across a variety of substrates, and provides a diverse range of heterocyclic products.
The metal-catalyzed, directing-group-promoted oxidative CH functionalization1 of aromatic C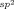 H bonds with alkynes2, 3 has been widely exploited to prepare a rich variety of heterocyclic4 and carbocyclic products.5 In the reactions of unsymmetrical alkynes, high regioselectivity is usually observed when the two substituents on the alkyne are electronically well-differentiated. For example, with alkynes containing one alkyl and one aryl substituent, the initial CC bond formation usually occurs with high regioselectivity at the alkyne carbon bearing the sp3-hybridized group. This regioselectivity is maintained in the oxidative annulation of 1,3-enynes, as demonstrated by the groups of Fagnou (Scheme 1 a)6a and Ackermann,6b for example. Herein, we describe a new mode of oxidative annulation, in which 1,3-enynes are able to function as one-carbon,7 rather than two-carbon reaction partners (Scheme 1 b). We propose this reactivity arises from a hitherto rare example of 1,4-RhIII migration, which opens up new possibilities in CH functionalization reactions.8 This phenomenon is general for substrates containing directing groups such as enols, phenols, carboxylic acids, or imides, resulting in a range of heterocyclic products.
H bonds with alkynes2, 3 has been widely exploited to prepare a rich variety of heterocyclic4 and carbocyclic products.5 In the reactions of unsymmetrical alkynes, high regioselectivity is usually observed when the two substituents on the alkyne are electronically well-differentiated. For example, with alkynes containing one alkyl and one aryl substituent, the initial CC bond formation usually occurs with high regioselectivity at the alkyne carbon bearing the sp3-hybridized group. This regioselectivity is maintained in the oxidative annulation of 1,3-enynes, as demonstrated by the groups of Fagnou (Scheme 1 a)6a and Ackermann,6b for example. Herein, we describe a new mode of oxidative annulation, in which 1,3-enynes are able to function as one-carbon,7 rather than two-carbon reaction partners (Scheme 1 b). We propose this reactivity arises from a hitherto rare example of 1,4-RhIII migration, which opens up new possibilities in CH functionalization reactions.8 This phenomenon is general for substrates containing directing groups such as enols, phenols, carboxylic acids, or imides, resulting in a range of heterocyclic products.

Oxidative annulation reactions of 1,3-enynes.
 (1)
(1)A possible mechanism for the formation of 4 a is shown in Scheme 2. Generation of the rhodium diacetate complex 5 from [{Cp*RhCl2}2] and Cu(OAc)2 is followed by cyclorhodation of substrate 1 a to provide the rhodacycle 6. Coordination and migratory insertion of the 1,3-enyne 2 a with the regioselectivity observed previously6 can then provide a new rhodacycle 7. Reductive elimination of 7 would then give the expected spiroindene 3 a as described with alkynes.5f,5g However, an alternative pathway is the reversible protonolysis of 7 with AcOH to provide the alkenylrhodium species 8, which can then undergo a 1,4-rhodium migration to give a new allylrhodium species 9 A.10 Notably, this process enables the activation of a C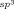 H bond. The 1,4-migration of rhodium(I) is well-known,11–13 but the corresponding 1,4-migrations of rhodium(III) are rare, with the only reports to date being stoichiometric studies of alkenyl to aryl migrations described by Ishii and co-workers.8 Presumably, the σ-allylrhodium species 9 A can interconvert with the π-allylrhodium species 9 B through the intermediacy of other isomers (not shown). Nucleophilic attack of the π-allylrhodium(III) moiety14, 15 of 9 B by the enol oxygen would provide the benzopyran 4 a and the rhodium(I) species 10, which can be then be reoxidized to 5 by Cu(OAc)2.
H bond. The 1,4-migration of rhodium(I) is well-known,11–13 but the corresponding 1,4-migrations of rhodium(III) are rare, with the only reports to date being stoichiometric studies of alkenyl to aryl migrations described by Ishii and co-workers.8 Presumably, the σ-allylrhodium species 9 A can interconvert with the π-allylrhodium species 9 B through the intermediacy of other isomers (not shown). Nucleophilic attack of the π-allylrhodium(III) moiety14, 15 of 9 B by the enol oxygen would provide the benzopyran 4 a and the rhodium(I) species 10, which can be then be reoxidized to 5 by Cu(OAc)2.

Proposed catalytic cycle.
A survey of reaction conditions16 revealed that lowering the temperature to 60 °C led to higher yields of benzopyran 4 a, but did not significantly alter the yield of spiroindene 3 a. Furthermore, the addition of AcOH (0.1 equiv) led to more consistently reproducible results. Under these conditions, benzopyran 4 a and spiroindene 3 a were obtained in 86 % and 12 % yield, respectively (Table 1, entry 1).
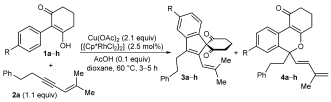
|
Entry |
Substrate |
Yield of 3 [%][b] |
Yield of 4 [%][b] |
|---|---|---|---|
|
1 |
1 a R=CO2Me |
12 |
86 |
|
2 |
1 b R=H |
60 |
20 |
|
3 |
1 c R=OMe |
64 |
17 |
|
4 |
1 d R=F |
26 |
44 |
|
5 |
1 e R=CF3 |
<5[c] |
78 |
|
6 |
1 f R=COMe |
<5[c] |
65 |
|
7 |
1 g R=NO2 |
n.d.[d] |
82 |
|
8 |
1 h R=SO2Me |
<5[c] |
84 |
- [a] Reactions were conducted with 0.50 mmol of 1 a–h. [b] Yield of isolated products. [c] The spiroindene was detected in trace amounts. [d] Not detected.
The scope of this transformation with respect to the 2-aryl-3-hydroxy-2-cyclohexenone was then explored (Table 1). With substrates 1 b and 1 c, which contain phenyl or 4-methoxyphenyl groups, respectively, the spiroindenes 3 were the major products (Table 1, entries 2 and 3). With substrates containing more electron-withdrawing substituents at the 4-position of the aromatic ring, the benzopyran became the major product (Table 1, entries 4–8). The spiroindene was formed in only trace amounts in the reactions of substrates containing trifluoromethyl, acetyl, or sulfone substituents (Table 1, entries 5, 6, and 8), and was not detected when a nitro group was present (Table 1, entry 7). These observations can be rationalized by considering that spiroindene formation requires the reductive elimination of RhIII from intermediates analogous to rhodacycle 7 (Scheme 2), with concomitant oxidation of the substrate. Therefore, it appears reasonable to assume that the activation barrier of this reductive elimination is increased with more electron-deficient substrates, as the substrate is more difficult to oxidize. The alternative pathway leading to the benzopyran 4 then becomes more competitive.
Next, the scope of this process with respect to the 1,3-enyne was investigated using substrate 1 g, and various enynes containing allylic hydrogens cis to the alkyne were shown to be effective one-carbon oxidative annulation partners (Table 2). None of the alternative spiroindenes were detected in any of these reactions. 1,3-Enynes containing protected or unprotected 2-hydroxyethyl groups were tolerated (Table 2, entries 1 and 2). 1,3-Enynes 2 d and 2 e, which contain a phenyl group or a hydrogen atom trans to the alkyne, also reacted smoothly to provide benzopyrans 11 d and 11 e (Table 2, entries 3 and 4). The reaction is not limited to 1,3-enynes containing methyl substitution cis to the alkyne, as shown by the successful annulations of 1,3-enynes 2 f and 2 g (Table 2, entries 5 and 6). Notably, a silyl-protected hydroxymethyl substituent at the trans-position of 1,3-enyne 2 h led to 11 h in 61 % yield with >95:5 E:Z selectivity at the enol silane (Table 2, entry 7).17 Finally, 1,3-enyne 2 h, which contains a methyl group at the alkenyl carbon proximal to the alkyne was also effective, providing 11 i with >95:5 E:Z selectivity (Table 2, entry 8).17

|
Entry |
1,3-Enyne |
Product |
Yield [%][b] |
|
|---|---|---|---|---|
|
1 2[c] |
2 b 2 c |
|
11 b R=OTBS 11 c R=OH |
84 62 |
|
3 4 |
2 d 2 e |
|
11 d R=Ph 11 e R=H |
73 64 |
|
5[d] 6 |
2 f 2 g |
|
11 f n=1 11 g n=2 |
93 89 |
|
7 |
2 h |
|
11 h |
61 |
|
8 |
2 i |
|
11 i |
61 |





- [a] Reactions were conducted with 0.50 mmol of 1 g. [b] Yield of isolated products. [c] Reaction conducted at 90 °C. [d] Reaction conducted using 0.37 mmol of 1 g.
This unusual oxidative annulation was found to be a general phenomenon, and not merely limited to 2-aryl-3-hydroxy-2-cyclohexenones. Several other aromatic substrates containing enol, phenol, carboxylic acid, or imide directing groups underwent oxidative annulation with 1,3-enyne 2 a to give a diverse range of five- or six-membered oxygen and nitrogen heterocycles 13 a–e (Scheme 3).18

Oxidative annulation reactions of various substrates with 1,3-enyne 2 a. Yields are of isolated products. [a] Reaction conducted in the presence of K2CO3 (3.0 equiv), and a second portion of [{Cp*RhCl2}2] (2.5 mol %) was added after 1 h. [b] Using 5 mol % of [{Cp*RhCl2}2].
 (2)
(2) (3)
(3) (4)
(4)
Possible mechanism of the 1,4-RhIII migration.
In conclusion, we have reported an unexpected mode of oxidative annulation in RhIII-catalyzed CH functionalizations when 1,3-enynes containing allylic hydrogens cis to the alkyne are present. The mechanism of these reactions is proposed to occur through double CH activation, including that of a C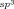 H bond, involving a hitherto rare example of the 1,4-migration of a RhIII species. Of broader significance, the generation of an allyl–metal species from sequential CH functionalization–1,4-metal migration opens up new opportunities in synthesis, and exploitation of this pathway in other transformations is underway in our laboratories.
H bond, involving a hitherto rare example of the 1,4-migration of a RhIII species. Of broader significance, the generation of an allyl–metal species from sequential CH functionalization–1,4-metal migration opens up new opportunities in synthesis, and exploitation of this pathway in other transformations is underway in our laboratories.

