

Hydrogen-Borrowing-Based Methods for the Construction of Quaternary Stereocentres
Abstract
Compounds containing quaternary stereocentres are a valuable motif in biologically active compounds. Herein we present our strategy to utilise the hydrogen borrowing manifold to access α-quaternary ketones via a tandem acceptorless dehydrogenation-cyclisation cascade. This new application of the methodology results in the formation of five- and six-membered carbocycles with a high degree of diastereoselectivity. Interestingly, benzylic alcohol substrates behaved anomalously and eliminated sulfinate in situ to give a set of rearranged α-quaternary ketone products.
Hydrogen borrowing (HB) catalysis is a powerful method for C−C bond formation using alcohols as alkylating agents.1 The method employs a transition metal catalyst which oxidises an alcohol to give a carbonyl compound and forms a metal hydride (hydrogen borrowing phase). This new carbonyl can undergo an aldol reaction to give an enone (condensation phase) which is reduced in situ by the aforementioned metal hydride (hydrogen returning phase). In this regard, we have shown that hindered Ph* (C6Me5) ketones are privileged substrates and can be alkylated using hydrogen borrowing to give both α- and β- branched (i. e. tertiary) products (Scheme 1A).2 Note that the mechanistic requirement for delivery of a hydrogen atom to both carbons of the enone during the hydrogen returning phase means that the formation of quaternary stereocentres cannot be accomplished directly using this method. This is unfortunate because the synthesis of stereochemically defined quaternary stereocentres is currently of high interest, especially in the design of biologically-relevant molecules which can benefit from their unique 3-dimensional properties.3 However, there is very little precedent for using hydrogen borrowing to form quaternary stereocentres at either the α- or β- positions relative to a carbonyl, with the work of Krische (using dienes & allenes)4 and Rodriguez (using organocatalysis/hydrogen borrowing) being the only related studies in this area.5 Therefore, we decided to design a general system that would exploit some steps of the hydrogen borrowing catalytic cycle, but would be modified to allow the formation of quaternary centres α- to a carbonyl group.

A) Previous work: Hydrogen borrowing for the formation of α- and β-branched carbonyls. B) This work: generation of α-quaternary stereocentres using an acceptorless dehydrogenation-cyclisation cascade. M=metal catalyst; Ph*=C6Me5.
Initially, we proposed that a quaternary stereocentre could be constructed by intercepting the unsaturated intermediate (made after in situ alcohol oxidation and condensation) with an appropriate nucleophile prior to the reduction step. In the full sequence, an alcohol substrate 1 (Scheme 1B), would be oxidised using a metal catalyst to give the aldehyde I, which could undergo condensation with an appropriate nucleophile 2 to yield electron-deficient alkene, II. If the reduction step of the HB catalytic cycle was inhibited, the pendant α-branched ketone enolate is poised to act as a nucleophilic trap to form a cyclic quaternary structure, such as 3. It was hypothesised that by employing an intramolecular enolate nucleophile to attack an unsaturated intermediate we could outcompete the undesired intermolecular alkene reduction process, as usually promoted by the metal hydride. This situation constitutes an acceptorless dehydrogenative coupling (or interrupted hydrogen borrowing) reaction.6, 7 It is important to note that the starting material, 1, can itself be made via hydrogen borrowing alkylation, enabling the construction of complex cyclic motifs using hydrogen borrowing methods in a short synthetic sequence. We aimed to study this system and prepare a range of carbocycles with quaternary centres, varying functionality and controlling stereochemistry.
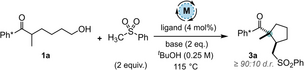
Our work began using substrate 1 a and PhSO2Me as the nucleophile (Table 1). Preliminary screening showed that two equivalents of base and sulfone were optimal for formation of the desired product 3 a at a reaction temperature of 115 °C (see ESI). Catalyst screening studies revealed that both [IrCp*Cl2]2 and Ru-MACHO® were poor catalysts for this reaction (notably, the Ru catalyst reduced the alkenyl sulfone intermediate (see II, Scheme 1) and produced undesired acyclic side product 4 in 55 % yield (Entries 1, 2). However, we did find that Ir(cod)acac was an effective catalyst in conjunction with a phosphine ligand,7 providing 3 a in up to 77 % isolated yield (Entries 3 and 4; 3 a was formed as a [93:7] mixture of diastereoisomers, vide infra). A commercially available manganese catalyst (Mn cat, Table 1) was also very effective for the transformation (Entry 5), giving product 3 a in 75 % yield.8 It was shown that the identity of the base had a profound effect on the yield (Entries 5–7), with KOtBu performing the best. Pleasingly, we were able to reduce the reaction time to 16 h (Entry 9) and isolated 3 a in 80 % yield; this reaction was also performed on a 4.00 mmol (1.10 g) scale to give product 3 a in 73 % yield and [94:6] d.r. (Entry 10). Finally, a control experiment conducted without the metal catalyst returned only starting material 1 a (Entry 11). The relative stereochemistry of 3 a was proven by both nOe analysis and X-ray crystallography,9 vide infra.
|
|||||
Entry |
[M] (mol%) |
Ligand |
Base |
t (h) |
Yield (%)[a] |
|---|---|---|---|---|---|
1 |
[IrCp*Cl2]2 (1) |
- |
KOtBu |
24 |
50 |
2 |
Ru-MACHO® (2) |
- |
KOtBu |
24 |
26 (28)[b] |
3 |
Ir(cod)acac (2) |
L1 |
KOtBu |
24 |
73 (77) |
4 |
Ir(cod)acac (2) |
L2 |
KOtBu |
24 |
71 |
5 |
Mn cat (2) |
- |
KOtBu |
24 |
74 (75) |
6 |
Mn cat (2) |
- |
NaOtBu |
24 |
17 |
7[c] |
Mn cat (2) |
- |
KOH |
24 |
57 |
8 |
Mn cat (2) |
- |
KOtBu |
4 |
55 |
9 |
Mn cat (2) |
- |
KOtBu |
16 |
77 (80) |
10 |
Mn cat (2) |
- |
KOtBu |
16 |
(73)[d] |
11 |
- |
- |
KOtBu |
24 |
0[e] |
|
|||||
- [a] Yields determined by 1H quantitative NMR spectroscopy using 1,1,2,2-tetrachloroethane as internal standard; isolated yields are given in parentheses. [b] compound 4 was isolated in 55 % yield. [c] PhMe (0.25 M) used as the reaction solvent [d] reaction performed on 4.00 mmol (1.10 g scale); 3 a was isolated in 94 : 6 d.r.; [e] Starting material 1 a was isolated in 79 % yield. Cp*=Me5C5; cod=1,5-cyclooctadiene; acac=acetylacetonate.
Using the optimised reaction conditions, we set about examining the scope of the transformation, concentrating on the earth abundant Mn-derived catalyst (Mn cat, Scheme 2).10 Electron deficient aryl sulfones were successful nucleophiles, giving 3 b, 3 c and 3 d in 48 %–72 % yield. Note that for substrates incorporating a more highly electron-withdrawing trifluoromethyl substituent, a competing SNAr process (see ESI) resulted in formation of the desired products in lower yields. Electron-rich aryl and heteroaryl sulfones were excellent substrates, giving 3 e and 3 f in 73 % and 69 %, respectively. A simple dialkyl sulfone was also found to be a good substrate, giving the desired product 3 g in 88 % yield. Given the success of using electron-rich aryl sulfones, we also investigated the use of sulfonamides as nucleophiles; these proved to be excellent substrates, giving the corresponding products 3 h, 3 i and 3 j in 77–82 % yield, respectively.

Scope of tandem acceptorless dehydrogenation-cyclisation cascade reaction. Reactions carried out on a 0.30 mmol scale. Single crystal X-ray structures are shown for the major diastereomer in all cases. Ph*=C6Me5, Ph×=2,3,5,6-C6Me4. [a] d.r. was determined post-purification. [b] 5 eq. nucleophile used. [c] Catalyst: Ir(cod)acac (4 mol%), CataCXium® A (L1,16 mol%). [d] Catalyst: Ir(cod)acac (2 mol%), CataCXium® A (4 mol%), Base: KOH (4 eq.), Solvent: PhMe (0.25 M).
Backbone substitution of keto-alcohol substrate 1 was also investigated. Pleasingly, larger ethyl, benzyl and benzyloxyethyl groups were tolerated at the quaternary position, giving 3 k, 3 l and 3 m in 56 %, 31 % and 25 % yields, respectively. Notably, the yield of cyclised product decreased as the size of the substituent increased, which is a testament to the challenge of synthesising such sterically encumbered motifs. Substitution could also be installed on the cyclopentane backbone itself, giving 3 n in 88 % yield (although here the level of diastereoselectivity was low). Pleasingly, cyclohexanes could also be prepared using this method using the homologous series of keto-alcohol starting materials, giving 3 o, 3 p, and 3 q in 72 %–96 % yield. In this case, high selectivity for the trans-substituted products was maintained, with the mono-substituted example 3 q showing improved diastereoselectivity, which we presume is reflective of the stronger conformational preferences of cyclohexanes compared to their cyclopentane analogues.11
Alternative nucleophiles to methyl sulfones were also explored and methyl ketones bearing a hindered aryl group, such as Ph*COMe, were tolerated, giving cyclopentanes 3 r and 3 s in 69 % and 83 % yield, respectively. Cyclopropyl methyl ketone was also an acceptable nucleophile, giving the diketone 3 t in 60 % yield. Acetonitrile was tolerated as a nucleophile, but gave a poor (15 %) yield of the cyclopentane product 3 u. Once again, cyclohexane analogues could be formed, as demonstrated by the synthesis of 3 v in 72 % yield; finally, bicyclic product 3 w was also prepared in 34 % yield using this method.
In each case we employed a Ph* ketone enolate as the nucleophilic trap (the main advantages of these ketones revolve around the fact that the C=O group is not electrophilic by virtue of the two shielding ortho methyl groups).12 Firstly, Ph* improves the yields dramatically, as shown by the fact that the Ph analogue of 1 a gave a complex mixture of products (for examples of unsuccessful substrates see ESI). Secondly, the Ph* group imparts crystallinity onto most compounds that incorporate it, allowing several single crystal X-ray structures to be obtained in order to prove the relative stereochemistry of the products.9
As shown in Scheme 2, the reaction demonstrated high levels of 1,2-trans-diastereoselectivity in the cyclisation. We suspected that this was a result of a thermodynamic preference during cyclisation, aided by the high steric demand of the Ph* group. To probe the implied reversible nature of the cyclisation, a small sample of the minor isomer of compound 3 a was isolated (cis-3 a). Upon re-subjection of cis-3 a to the basic reaction conditions, 3 a was recovered in the same ratio as the original cyclisation ([93:7] d.r.), with trans-3 a as the major diastereomer. This experiment demonstrates that the cyclisation is reversible and the product d.r. is controlled by the relative stabilities of the diastereomeric products.13
An important property of the key Ph* protecting group is that it can be readily cleaved to give a range of different carbonyl derivatives (Scheme 3).2, 12 As such, the Ph* group was removed from the cyclic products via a retro-Friedel Crafts acylation reaction using either acid or Br2 to form an acylium ion. This intermediate could then be intercepted in situ by a range of nucleophiles to give products 5 – 10 in high yields and with complete retention of relative stereochemistry, as proven by nOe and single crystal X-ray structure analysis (see ESI).

Derivatisation of the Ph* group; all products are [>95:5] d.r. (a) HCl (2 M), HFIP, 65 °C, 16 h. For all other examples: Br2 (2 eq.) CH2Cl2, −17 °C, then add (b) LiAlH4 (5 eq.); (c) BuOH (3 eq.); (d) 3,5-ditrifluoromethyl benzylamine (2 eq.), iPr2NEt (4 eq.) (e) N-hydroxy phthalimide (2 eq.), iPr2NEt (4 eq.); (f) NaHCO3 (aq.).
While investigating the role of unsaturation on the substrate backbone, we discovered some unexpected reactivity involving sulfone nucleophiles. Our initial investigations with 11 (Scheme 4, R=Me) revealed that two distinct products (12 a and 13 a) were formed, and the ratio between them could be altered by changing the catalyst used. After optimisation (see ESI), we discovered that a Ru-MACHO® catalyst was the most effective for the formation of exocyclic alkene 12, while [IrCp*Cl2]2 was optimal for the formation of six-membered ring product 13. Under the Ru-MACHO® conditions, a range of groups were tolerated at the quaternary position, giving 12 a, 12 b, and 12 c in 85 %, 76 % and 51 % yields, respectively. Substituents were also tolerated on the aromatic ring in the substrate backbone, giving 12 d–12 g in 42 %–92 % yield. An electron deficient benzylic alcohol also cyclised to give 12 h in moderate 43 % yield. Unfortunately, the scope for the Ir-catalysed reaction to form 6-membered carbocycles was more limited. While the simple 6-membered ring product 13 a could be isolated in 70 % yield, the corresponding ethyl-substituted product 13 b was formed in much lower yield. Here, only simple substitution was tolerated on the backbone aromatic ring to give 13 c in 67 % yield.

Divergent behaviour of benzylic alcohol substrates. Reactions carried out on 0.30 mmol scale unless otherwise indicated. [a] Reaction carried out on 0.20 mmol scale. [b] Reaction carried out on 0.15 mmol scale. [c] Yield determined via 1H quantitative NMR using 1,1,2,2-tetrachloroethane as an internal standard.
Next, we studied the mechanism by which the products 12 and 13 were formed, and suggest III (formed after alcohol oxidation and condensation with the sulfone) is a key intermediate (Scheme 5A). We propose that the relative rates by which III is either reduced or cyclises (and eliminates) governs the product distribution. When using Ru-MACHO® reduction of the alkene in III is slow relative to cyclisation and pathway (i) is followed. This pathway involves deprotonation adjacent to the ketone and 5-exo-trig cyclisation to form IV. Elimination of sulfinate, expedited by the acidifying effect of the aromatic ring, results in the formation of exocyclic alkene 12.14 Supporting this hypothesis is the observation that subjection of putative aldehyde intermediate 14 to the reaction conditions, in the absence of metal catalyst, furnished alkene 12 a in excellent yield; this indicates that the metal is only involved in the oxidation phase of this reaction (Scheme 5B).

A) Mechanistic proposal for the divergent behaviour of benzylic alcohol substrates. (i) Cyclisation and then elimination; (ii) reduction, elimination and then cyclisation. B) Supporting mechanistic experiments
However, when using [IrCp*Cl2]2 we suggest that reduction of the alkene in III is fast relative to cyclisation and so intermediate V will be formed and hydrogen borrowing pathway (ii) followed (Scheme 5A). Next, elimination of sulfinate (again aided by the aromatic ring) would provide styrene VI,14 which could undergo enolate formation and 6-endo-trig cyclisation. This hypothesis is supported by the observation that when styrene 15 was subjected to the reaction conditions (again in the absence of metal catalyst) then 6-endo-trig cyclisation produced 13 a in 70 % yield.
The preference shown for initial reduction of III with [Ir] and initial cyclisation/elimination of III with [Ru] is complex because it may be related to the rates of alkene reduction and/or the rates by which the catalysts evolve hydrogen and so prevent reduction.8, 13 In this regard, we note that Ru-MACHO-type complexes are often employed in acceptorless dehydrogenation.6
Finally, we conducted a brief investigation of the reactivity of the cyclic products thus prepared (Scheme 6). While cleavage of the Ph* group from product 13 a was very high yielding, giving the acid fragment 16 in 99 % yield, the Ph* group could not be removed from unsaturated product 12 a due to unwanted reactivity of the alkene. However, exocyclic alkene 12 a could be reduced from either face using hydrogenation under steric (17) or chelation (18) control. As expected, the Ph* group could be readily cleaved to form carboxylic acids 19 and 20 in a single step. Moreover, acid 20 was subsequently coupled to 2-aminothiazole to prepare a known biologically active derivative, 21 in moderate yield.15

A) Ph* cleavage of 6-membered ring products. B) Derivatisation of 5-membered ring products 12 a and 13 a via Ph* cleavage.
To conclude, we have developed conditions for using acceptorless dehydrogenation and hydrogen borrowing methods for the formation of functionalised and stereochemically defined carbocycles. By exploiting the electrophilic nature of a Michael acceptor intermediate within the HB catalytic cycle we have been able to employ a subsequent cyclisation step to generate a quaternary centre and have been able to overcome one of the major limitations of hydrogen borrowing methodology. The key Ph* ketone protecting group was easily cleaved to form a range of useful complex fragments bearing quaternary stereocentres.
Acknowledgments
J.L.C. is grateful to the Centre for Doctoral Training in Synthesis for Biology and Medicine for a studentship, generously supported by GSK, MSD, Syngenta and Vertex, to the Royal Commission for the Exhibition of 1851 for an Industrial Fellowship and to the Engineering and Physical Sciences Research Council [EPSRC Doctoral Training Partnership, Excellence Award (EP/T517811/1)].
Conflict of Interests
The authors declare no conflict of interest.

