

Enantio- und regiokonvergente, nickelkatalysierte Veretherung von Phenolen durch Allylierung zur Darstellung chiraler C(sp3)−O-Allylarylether
Abstract
Es wird eine enantio- und regiokonvergente Allylierung von Phenolen unter Nickelkatalyse mit einem α-/γ-Regioisomerengemisch racemischer silylierter/germylierter Allylchloride beschrieben. Die Silyl-/Germylgruppe steuert die Regioselektivität, und die Umwandlung liefert enantiomerenangereicherte unsymmetrische 1,3-disubstituierte Allylarylether mit ausgezeichneter Regiokontrolle in guten Ausbeuten und mit hervorragenden Enantioselektivitäten. Bemerkenswerterweise wird keine nickelvermittelte C−O-Bindungsaktivierung bei Raumtemperatur beobachtet. Der synthetische Wert dieser hochfunktionalisierten, siliciumhaltigen Bausteine wird in einer Reihe chemoselektiver Umwandlungen aufgezeigt, einschließlich einer [3,3]-sigmatropen Umlagerung zur Herstellung eines α-chiralen Silans.
Sauerstofftragende stereogene Kohlenstoffatome sind allgegenwärtig in der Synthesechemie, und chirale Allylarylether sind wichtige Vertreter dieser Verbindungsklasse.1, 2 Ihre Synthese durch SN2- oder sogar SN1-Reaktionen von chiralen allylischen Elektrophilen ist mit Problemen der Regioselektivität und dem Verlust stereochemischer Informationen verbunden. In den letzten zwei Jahrzehnten wurden übergangsmetallkatalysierte Verfahren entwickelt, um diese Herausforderungen zu überwinden, wobei die meisten auf allylischen Substitutionen ausgehend von verzweigten oder linearen allylischen Elektrophilen unter Durchlaufen terminaler π-Allyl-Zwischenstufen von Ruthenium,3 Rhodium,4 Palladium5, 6 und Iridium6a, 7, 8 mit Sauerstoffnukleophilen beruhen (Schema 1, oben). Alternativ können verzweigte allylische Ether mit hoher Enantioselektivität durch eine elegante Palladium(II)-katalysierte SN2’-Reaktion von allylischen Trichloracetimidaten hergestellt werden.9, 10 Darüber hinaus ist die Verwendung von acyclischen symmetrischen 1,3-disubstituierten allylischen Elektrophilen zur Veretherung mit Palladium als bewährtem Katalysator gut etabliert (Schema 1, Mitte).6, 11 Im Gegensatz dazu bleibt die Kontrolle über die Regio- und Enantioselektivität sowie die Alkengeometrie für unsymmetrische 1,3-disubstituierte allylische Ether eine Herausforderung und ist bislang selten beschrieben worden.12, 13

Frühere Berichte über asymmetrische allylische Substitutionsreaktionen zur Herstellung chiraler allylischer C(sp3)−O-Einheiten. Alk=Alkyl, Ar=Aryl.
Unsere Arbeitsgruppe hatte kürzlich gezeigt, dass sterisch anspruchsvolle Silyl-14 und Germylgruppen15 in allylischen Elektrophilen die nickelkatalysierte C(sp3)−C(sp3)-Bindungsknüpfung vom silicium- oder germaniumsubstituierten Kohlenstoffatom weglenken können und so ein einziges Regioisomer bilden. Auf dieser Grundlage wollten wir diese Strategie nutzen, um eine stereokonvergente Umwandlung von unsymmetrischen 1,3-disubstituierten allylischen Elektrophilen in chirale Allylarylether zu verwirklichen (Schema 1, unten).
An dieser Stelle ist es wichtig zu erwähnen, dass Takeda und Mitarbeiter vor fast zwanzig Jahren über ein racemisches Verfahren für die Synthese von allylischen Ethern und Thioethern unter Nickelkatalyse berichteten.16 Dieses alleinstehende Beispiel ist nicht nur für die nickelkatalysierte O-Allylierung wegweisend, sondern auch bedeutend, weil es belegt, dass bei einer Reaktionstemperatur von 50 °C keine konkurrierende, nickelvermittelte C−O-Bindungsaktivierung auftritt.17, 18, 19, 20 Die Entwicklung katalytischer Prozesse mit Nickel als häufig vorkommendes Element ist besonders wünschenswert,21 und hier berichten wir über eine hochgradig enantioselektive allylische Substitution mit Phenolnukleophilen, die mit hervorragender Regiokontrolle verläuft.
Wir begannen unsere Untersuchung mit dem Me2PhSi-substituiertem Allylchlorid rac-1 a als Modellsubstrat in Gegenwart von Phenol, einer Base und einem Nickelkatalysator (Tabelle 1). Ein Screening von Lösungsmitteln zeigte frühzeitig, dass in polaren aprotischen Lösungsmitteln wie DMA oder DMF Natriumphenolat oder Phenol/Base-Kombinationen direkt mit dem allylischen Elektrophil reagieren. Ohne Beteiligung des chiralen Nickelkatalysators wurde keine Enantioinduktion, jedoch eine gute Kontrolle der Regioselektivität bei der Bildung von Produkt 3 aa beobachtet. Die konkurrierende SN2-Reaktion wurde in Toluol vollständig unterdrückt, und die nickelkatalysierte allylische Substitution mit dem chiralen Box-Liganden L1 ermöglichte die Bildung von Produkt 3 aa in geringer Ausbeute, aber mit ausgezeichneter Enantioselektivität als einzelnes Regioisomer (Nr. 1). Andere chirale Liganden lieferten keine weiteren Verbesserungen (für die vollständige Liste der getesteten Liganden siehe die Hintergrundinformationen). Zum Beispiel führte PyBox L2 zu einer deutlich höheren Ausbeute, das aber mit geringerer Enantioselektivität (Nr. 2), und die Verwendung von QuinOx L3 führte zu keiner Produktbildung (Nr. 3). Im Gegenzug hatte die Base einen erheblichen Einfluss auf die Ausbeute und das unter Beibehaltung des hohen Niveaus der Enantioselektivität (Nr. 4–6). Die Verwendung von Cs2CO3 anstatt K3PO4 verbesserte zwar die Ausbeute, aber NaH brachte das beste Ergebnis, und es wurde eine Ausbeute von 71 % mit >99 % ee für 3 aa erzielt; eine organische Base wie DBU war im Vergleich zu den anorganischen Basen unterlegen.
|
|||
Nr. |
Abweichung von den Standardbedingungen |
Ausbeute [%][b] |
ee [%][c] |
|---|---|---|---|
1 |
keine |
23 |
98 |
2 |
L2 anstelle von L1 |
74 |
79 |
3 |
L3 anstelle von L1 |
Spuren |
n.b. |
4 |
Cs2CO3 anstelle von K3PO4 |
39 |
98 |
5[d] |
NaH anstelle von K3PO4 |
71 |
>99 |
6 |
DBU anstelle von K3PO4 |
11 |
98 |
7[d] |
THF anstelle von Toluol |
10 |
98 |
8[d] |
2-Me-THF anstelle von Toluol |
64 |
90 |
9[d] |
(Ph3P)4Ni anstelle von Ni(cod)2 |
k.U. |
n.b. |
10[d] |
NiCl2/Mn (3.0 Äquiv.) anstelle von Ni(cod)2 |
k.U. |
n.b. |
11[d] |
50 °C anstelle von RT |
23 |
99 |
12[d] |
mit 15-Krone-5 (1.2 Äquiv.) |
61 |
90 |
13[d] |
NaOPh (1.2 Äquiv.) anstelle von PhOH/NaH |
30 |
98 |
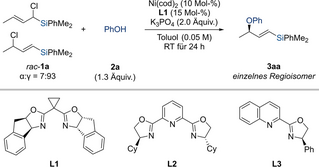
- [a] Alle Reaktionen wurden im 0.10-mmol-Maßstab durchgeführt. [b] Bestimmt mittels GLC-Analyse mit Biphenyl als internem Standard. [c] Bestimmt mittels HPLC-Analyse an einer chiralen stationären Phase. [d] 1.3 Äquiv. an NaH als Base benutzt und 12 h Reaktionszeit. n.b.=nicht bestimmt. k.U.=kein Umsatz. DBU=1,8-Diazabicyclo[5.4.0]undec-7-en. 15-Krone-5=1,4,7,10,13-Pentaoxacyclopentadecan.
Wir fuhren mit der Abklärung weiterer Parameter fort (für den vollständigen Datensatz siehe die Hintergrundinformationen). Selbst mit NaH als Base war Toluol eine bessere Wahl als THF und 2-Me-THF (Nr. 7 und 8). Mit (Ph3P)4Ni oder NiCl2/Mn als Nickelpräkatalysator fand keine Reaktion statt (Nr. 9 und 10). Weder die Erhöhung der Reaktionstemperatur noch die Zugabe von 15-Krone-5 hatten einen vorteilhaften Einfluss (Nr. 11 und 12). Der hohe ee-Wert wurde mit Natriumphenolat beibehalten, jedoch mit geringerer Ausbeute (Nr. 13). Wir vermuten, dass das Phenolat Spuren von Wasser enthält, und da ein Kontrollexperiment mit drei Äquivalenten Wasser keine Produktbildung zeigte, könnte das erklären, warum die Effizienz höher ist, wenn das Phenolat lokal erzeugt wird.
Da weitere Optimierungen der Reaktionsbedingungen unter Nr. 5 ergebnislos blieben, wandten wir uns der Untersuchung der Anwendungsbreite zu (Schema 2). Der Einfluss des Substitutionsmusters am Siliciumatom zeigte keinen eindeutigen Trend. Gute Ausbeuten und hervorragende Enantioselektivitäten wurden mit Trialkylsilylgruppen wie Et3Si- in 1 b, tBuMe2Si- in 1 c und BnMe2Si- in 1 d erzielt; das allylische Elektrophil mit der sterisch sehr anspruchsvollen tBu2MeSi-Gruppe ergab nur Spuren des Produkts (nicht gezeigt). Arylgruppen am Siliciumatom waren kompatibel, wie das Modellsubstrat 1 a mit Me2PhSi- und das verwandte 1 e mit MePh2Si- zeigten. Die sterisch stärker gehinderte Ph3Si-Gruppe in 1 f lieferte eine niedrigere Ausbeute sowie einen deutlich verringerten ee-Wert, was eine Einschränkung der Methodik aufzeigt.

Anwendungsbreite I: Variation der Silyl- und Germylgruppe und des Alkylsubstituenten. Alle Reaktionen wurden im 0.10-mmol-Maßstab durchgeführt. Die isolierten Ausbeuten beziehen sich auf analytisch reine Produkte nach Flashchromatographie an Kieselgel; die Ausbeuten in Klammern beziehen sich auf den Umsatz. Die Enantiomerenüberschüsse wurden mittels HPLC-Analyse an chiralen stationären Phasen ermittelt. [a] Eine Ausbeute von 51 % mit 96 % ee wurde in einem 1.0-mmol-Maßstab erhalten.
Ein Experiment im 1.0-mmol-Maßstab wurde mit dem Modellsubstrat durchgeführt, bei dem es zu leichten Abnahmen der Ausbeute und der Enantioselektivität kam (1 a→3aa). Verlängerungen der aliphatischen R’-Gruppe im Elektrophil wurden eingeführt und lieferten die chiralen allylischen Ether in hervorragenden Enantioselektivitäten als einzelne Regioisomere (1 g→3ga und 1 h→3ha). Eine Phenylgruppe am Ende der linearen Alkylkette war je nach Entfernung zum Reaktionszentrum kompatibel; Homobenzyl zeigte gute Ergebnisse (1 j→3ja), Benzyl jedoch nicht, wobei sowohl die Ausbeute als auch der ee-Wert deutlich abnahmen (1 i→3ia). Dies führen wir auf sterische Hinderung zurück, was in Übereinstimmung mit der experimentellen Feststellung steht, dass die allylische Substitution durch eine Isopropylgruppe in dieser Position behindert wird (nicht gezeigt). Zum Schluss wurde gezeigt, dass die Reaktionsvorschrift auch auf die entsprechenden Germaniumderivate anwendbar ist (4 a→5aa und 4 b→5ba).
Als Nächstes untersuchten wir das Substitutionsmuster des Phenolnukleophils in der Modellreaktion (2 b–u; Schema 3). Die drei regioisomeren Kresole 2 b–d ergaben die Produkte 3 ab–ad mit einem Enantiomerenüberschuss von 90 % oder höher; die Ausbeute und Enantioinduktion waren beim o-Kresol (1 d) am niedrigsten und beim m-Kresol (1 c) am höchsten. Dieses Ergebnis zeigte sich auch bei anderen Phenolderivaten, die zwei oder einen Substituenten in der ortho-Position tragen (2 e→3ae sowie 2 o→3ao und 2 u→3au); im Fall des Produkts 3 ae wurde das α-Regioisomer festgestellt, was erneut darauf hindeutet, dass es Einschränkungen für sterisch anspruchsvolle Reaktanden gibt. Andere funktionelle Gruppen, die in der para-Position toleriert werden, wie tert-Butyl (2 f), Phenyl (2 g), Methoxy (2 i), ein Acetal (2 k), ein Thioether (2 l), eine Fluorgruppe (2 m) und ein Boronat (2 n) lieferten die allylischen Ether in guten Ausbeuten und mit hohen Enantioselektivitäten. Darüber hinaus sind β-Naphthol (2 h), 3,5-Dimethoxyphenol (2 j) und ein ketonhaltiges Phenol (2 p) vollständig mit den allgemeinen Bedingungen vereinbar. Wir mussten feststellen, dass elektronenarme Phenolderivate mit einer Cyano- oder Carboxylgruppe in der para-Position nicht umgesetzt werden (nicht gezeigt). Natürlich vorkommende Phenole sowie eine biologisch aktive Substanz lieferten die O-allylierten Produkte in guten Ausbeuten und mit erstklassigen Enantioselektivitäten (2 r–t→3ar–at und 2 q→3aq).

Anwendungsbreite II: Variation des Sauerstoffnukleophils. Alle Reaktionen wurden mit 0.10-mmol Allylchlorid rac-1 a, 1.3 Äquiv. mit den Phenolen 2 b–u, 10 Mol-% an Ni(cod)2, 15 Mol-% an L1, und 1.3 Äquiv. an NaH in Toluol (2.0 mL) bei Raumtemperatur für 12 Stunden durchgeführt. Die isolierten Ausbeuten beziehen sich auf analytisch reine Produkte nach Flashchromatographie an Kieselgel; die Ausbeuten in Klammern beziehen sich auf den Umsatz. Die Enantiomerenüberschüsse wurden mittels HPLC-Analyse an chiralen stationären Phasen ermittelt. [a] r.r.=93 : 7. [b] d.r.=50 : 50. r.r.=Regioisomerenverhältnis.
Um die entscheidende Rolle der Silylgruppe bei der Steuerung der C(sp3)−O-Bindungsbildung zu veranschaulichen, führten wir zwei Kontrollexperimente durch (Schema 4A). Zunächst lieferte rac-6 a mit zwei linearen Alkylketten den allylischen Ether 7 als Regioisomerengemisch mit schlechter Enantioselektivität. Nach Austausch einer Alkylkette gegen eine Arylgruppe führten wir eine weitere Reaktion mit Cinnamylchloridderivat rac-8 a durch, welches wiederum das Produkt mit geringer Regioselektivität und Enantioselektivität lieferte. Im Hinblick auf die Fähigkeit von Nickel C−O-Bindungen zu aktivieren,17, 18, 19, 20 führten wir ein Crossoverexperiment durch, um die Reversibilität der C(sp3)−O-Bindungsbildung zu untersuchen. Als das Produkt 3 aa den Standardbedingungen in Gegenwart von p-Methoxyphenol (2 i) unterworfen wurde, wurde kein Crossoverprodukt 3 ai nachgewiesen; der allylische Ether 3 aa konnte ohne Verlust der stereochemischen Information zurückgewonnen werden (Schema 4B). Dieses Ergebnis schließt jegliche Reversibilität der C(sp3)−O-Bindungsbildung aus. Das Abfangen von einem möglichen Radikalintermediat wurde mit der Zugabe von 2,2,6,6-Tetramethylpiperidin-1-oxyl (TEMPO) als Radikalfänger unter den Standardreaktionsbedingungen durchgeführt, wobei es zu keiner Produktbildung kam; die Bildung des entsprechenden TEMPO-Addukts wurde mittels HRMS-Analyse bestätigt (Schema 4C). Hinweis: Ein Substrat, welches eine rasche radikalische Ringöffnung eingehen könnte, konnte aufgrund seiner chemischen Instabilität nicht isoliert werden. Aufbauend auf diesen Kontrollexperimenten schlagen wir die Bildung eines Allylradikals vor, was auch die Stereokonvergenz erklärt (für den vorgeschlagenen Katalysecyclus und ein stereochemisches Modell siehe die Hintergrundinformationen).22

Mechanistische Untersuchungen. Alle Reaktionen wurden im 0.10-mmol-Maßstab durchgeführt. Die isolierten Ausbeuten beziehen sich auf analytisch reine Produkte nach Flashchromatographie an Kieselgel. Die Enantiomerenüberschüsse wurden mittels HPLC-Analyse an chiralen stationären Phasen ermittelt. PMP=p-Methoxyphenyl.
Die hochgradig funktionalisierten Allylether 3 aa und 3 ai bieten etliche Möglichkeiten für weiterführende chemoselektive Umwandlungen (Schema 5). Die Spaltung des Arylethers entspricht einer Entschützung eines allylischen Alkohols. Die Ein-Elektronen-Oxidation zur Entfernung der PMP-Gruppe in 3 ai lieferte den literaturbekannten Allylalkohol 10 in 86 % Ausbeute ohne jegliche Racemisierung. Durch Vergleich der optischen Drehung mit dem berichteten Wert23 wurde die absolute Konfiguration als R zugeordnet (siehe die Hintergrundinformationen für Details). Weitere Umsetzungen wurden mit dem Allylether 3 aa durchgeführt. Durch das Nutzen der Vinylsilaneinheit als Ausgangspunkt konnten sowohl eine Hiyama-Kreuzkupplung zur Bildung von 11 als auch eine Halodesilylierung zur Erzeugung von 12 in moderaten Ausbeuten verwirklicht werden. Bei der Verwendung von ICl anstelle von NIS stellten wir fest, dass die Addition an die C−C-Doppelbindung und nicht der Si−I-Austausch stattfindet. Das hochfunktionalisierte Produkt 13 wurde in guter Ausbeute und ohne Verlust der stereochemischen Information erhalten. Außerdem gelang uns die racemierungsfreie Hydrierung des Vinylsilans 3 aa zum Alkylsilan 14 in 62 % Ausbeute. Der nachfolgende oxidative Abbau der C(sp3)−Si-Bindung in 14 nach Fleming24 ermöglichte den Zugang zu dem literaturbekannten Alkohol 15 in 95 % Ausbeute. Die erwartete Absolutkonfiguration des Produkts 15 steht im Einklang mit der Literatur.25 Zuletzt ermöglichte eine Lewis-Säure-katalysierte Claisen-Umlagerung die Umwandlung des chiralen Allylarylethers 3 aa in das α-chirale Silan 16 und damit einen erfolgreichen 1,3-Chiralitätstransfer mit der gewünschten C−C-Bindungsbildung mit geringfügiger Verringerung der Enantioselektivität; die Doppelbindungsgeometrie konnte nicht kontrolliert werden.11b

Derivatisierung der Produkte. Die isolierten Ausbeuten beziehen sich auf analytisch reine Produkte nach Flashchromatographie an Kieselgel. Die Enantiomerenüberschüsse wurden mittels HPLC-Analyse an chiralen stationären Phasen ermittelt. [a] Die relative Stereochemie dieses acyclischen Moleküls konnte nicht zugeordnet werden. [b] CAN (2.0 Äquiv.), MeCN/H2O (3 : 1, 0.10 M), RT für 12 h. [c] (Ph3P)2PdCl2 (5.0 Mol-%), Ph3P (10 Mol-%), CuI (1.0 Äquiv.), TBAF (3.0 Äquiv.), PhI (1.2 Äquiv.), DMF (0.10 M), 100 °C für 15 h. [d] NIS (3.0 Äquiv.), MeCN (0.10 M), 60 °C für 15 h. [e] ICl (1.0 Äquiv.), CH2Cl2 (0.10 M), 0 °C bis RT für 1 h. [f] Pd/C (2.0 Mol-%), H2 (1 atm), MeOH (0.10 M), RT für 12 h. [g] Hg(OAc)2 (1.5 Äquiv.), CH3CO3H (35 % wt in AcOH, 1.0 mL), RT für 12 h. CAN=Cerammoniumnitrat, NIS=N-Iodsuccinimid, TBAF=Tetra-n-butylammoniumfluorid.
Wir stellten hier eine effiziente enantio- und regiokonvergente Veretherung regioisomerer Gemische von silylierten und germylierten Allylchloriden mit Phenolnukleophilen unter Nickelkatalyse vor. Die Metalloidgruppe ist erneut der Schlüssel zum Erfolg, da sie die Regioselektivität steuert und die unsymmetrischen 1,3-disubstituierten chiralen Allylether als einzelnes Regioisomer liefert. Darüber hinaus sind allgemeine Methoden zur Erzeugung von C−O-Bindungen unter Nickelkatalyse bislang kaum entwickelt, möglicherweise auch, weil Nickel dafür bekannt ist, solche Bindungen zu aktivieren.17, 18, 19, 20 Der synthetische Wert der Produkte wurde durch die Entfernung der PMP-Schutzgruppe zur Freisetzung des entsprechenden Allylalkohols und durch andere chemoselektive Umwandlungen veranschaulicht.
Danksagung
Diese Arbeit wurde von der Deutschen Forschungsgemeinschaft (Oe 249/25-1) und dem Land Berlin (Elsa-Neumann-Stipendium zur Vorbereitung der Promotion für D.N., 2023–2025) gefördert. M.O. ist der Einstein Stiftung Berlin für eine Stiftungsprofessur zu Dank verpflichtet. Open Access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.
Interessenkonflikt
Die Autoren erklären, dass keine Interessenkonflikte vorliegen.
Open Research
Data Availability Statement
Die Daten, die die Ergebnisse dieser Studie unterstützen, sind auf begründete Anfrage beim Autor erhältlich.

