

Mesoionische N-Heterocyclische Olefine verschieben die obere Grenze der Nucleophilie-Skala
Abstract
Mesoionische, 1,2,3-Triazol-abgeleitete N-heterocyclische Olefine (mNHOs) mit außerordentlich elektronenreicher, exocyclischer CC–Doppelbindung wurden synthetisiert und spektroskopisch charakterisiert, z. T. durch Röntgenstrukturanalyse. Die Kinetik der mNHO-Reaktionen mit Arylidenmalonaten (ArCH=C(CO2Et)2) zu zwitterionischen Addukten wurde photometrisch in THF bei 20 °C verfolgt. Die resultierenden Geschwindigkeitskonstanten 2. Ordnung k2(20 °C) korrelieren linear mit zuvor bestimmten Elektrophilieparametern E der Arylidenmalonate (Referenzelektrophile) und liefern gemäß der Beziehung lg k2(20 °C)=sN(N+E) die nucleophilspezifischen N- und sN-Parameter der mNHOs. Die mNHOs sind mit 21<N<32 viel stärkere Nucleophile als herkömmliche NHOs. Einige mNHOs übertreffen sogar die Reaktivität von mono- und diakzeptor-substituierten Carbanionen. Es wird an Beispielen gezeigt, dass es die so bestimmten Reaktivitätsparameter ermöglichen, die Geschwindigkeitskonstanten für mNHO-Reaktionen mit weiteren Michael-Akzeptoren zu berechnen. Auch die Reaktionen mit anderen elektrophilen Reaktionspartnern, inklusive Kohlenstoffdioxid, das zwitterionische mNHO-Carboxylate bildet, lassen sich vorhersagen. Die Nucleophilie-Parameter N korrelieren linear mit einer Linearkombination der quantenchemisch berechneten Methylkation-Affinitäten und %Vbur („buried volumes“) der mNHOs, was ein wertvolles Werkzeug liefert, um Reaktivitäten starker Kohlenstoff-Nucleophile maßzuschneidern.
Einleitung
Die Reaktivität von olefinischen π-Systemen wird stark durch elektronenschiebende bzw. -ziehende Substituenten beeinflusst. Ein Konzept zur Erzeugung starker, neutraler Kohlenstoff-Nucleophile ist die ylidische Polarisierung von π-Systemen durch geminale Substitution mit Donorgruppen. In Keten-N,N-acetalen (1,1-Endiaminen) A1,1 beispielsweise, erhöhen zwei Dialkyl-aminogruppen die Elektronendichte der olefinischen π-Bindung (Schema 1). Die elektronendonierende Wirkung der beiden Aminogruppen kann noch verstärkt werden, indem sie in konformationell starre Heterocyclen (A2) eingebettet werden (Schema 1).2, 3 Solche stark polarisierten N-heterocyclischen Olefine (NHOs) können durch eine neutrale (A2) oder eine zwitterionische Lewis-Struktur (A2′) dargestellt werden, bei der die exocyclische Methyleneinheit eine negative Ladung trägt. NHOs fanden aufgrund ihrer starken Donoreigenschaften4 zahlreiche Anwendungen, die von Organokatalyse5 über Hauptgruppenchemie6 bis hin zu Polymerchemie reichen.7 Basizität und Nucleophilie von NHOs sind bemerkenswert hoch und übertreffen in vielen Fällen die von N-heterocyclischen Carbenen (NHCs).8

Strukturelle Evolution starker Donoren mit Kohlenstoffzentrum: Von acyclischen 1,1-Diaminoolefinen über NHOs zu mNHOs (Dipp=2,6-Diisopropylphenyl).
2020 stellte die Hansmann-Gruppe ein neues Konzept zur Erzeugung starker Kohlenstoffdonoren vor, das auf mesoionischer Kohlenstoff-Kohlenstoff-Bindungspolarisation beruht.9 Solche mesoionischen Methylide oder mesoionischen N-heterocyclischen Olefine (mNHOs) auf der Basis von 4-Methylen-1,3-imidazolen oder 4-Methylen-1,2,3-triazolen (A3) können nicht durch neutrale, sondern nur durch eine Reihe zwitterionischer Lewis-Strukturen beschrieben werden (Schema 1).10, 11 Konkurrenzexperimente und Tolmans elektronische Parameter (TEP) zeigten, dass die Donoreigenschaften der exocyclischen Methylengruppen in mNHOs die der analogen =CH2-Gruppe in klassischen NHOs übertreffen.9
Schon bald nach der ersten Veröffentlichung wurden die neuen mNHOs A3 und analoge Imidazolderivate zur Synthese raumtemperaturstabiler Diazoalkene durch Reaktion mit N2O,12 in der Organokatalyse13 und in der Koordinationschemie eingesetzt.14 Darüber hinaus wurde berichtet, dass die von Triazolen abgeleiteten mNHOs (4+1)-Cycloadditionsreaktionen mit Diazoestern15 eingehen. Quantenchemische (DFT−)Berechnungen sagen zudem voraus, dass mNHOs geeignet sein könnten, das Treibhausgas SF6 zu zersetzen.16
 (1)
(1)In der linearen Freie-Energie-Beziehung (1) werden Elektrophile durch einen Elektrophilieparameter E charakterisiert. Die Reaktivität von Nucleophilen wird durch zwei lösungsmittelabhängige Parameter beschrieben, die Nucleophilie N und die Suszeptibilität sN.17, 18 Mittels Gleichung (1) lassen sich die Geschwindigkeitskonstanten 2. Ordnung für Reaktionen von Nucleophilen mit Elektrophilen in der Regel mit einer Genauigkeit von zwei Größenordnungen innerhalb eines Reaktivitätsbereich von 40 Größenordnungen vorhersagen.17d Das Ziel dieser Arbeit war es, das synthetische Potenzial von mNHOs zu erkunden, d. h., potenzielle elektrophile Reaktionspartner effizient zu identifizieren und ein Fundament für die Entwicklung neuartiger mNHOs mit spezifischen Reaktivitäten zu schaffen. Hierzu bestimmten wir die N- und sN-Parameter der mNHOs, indem wir die Kinetik ihrer Reaktionen mit etablierten Referenzelektrophilen untersuchten.
Ergebnisse und Diskussion
Synthese und Charakterisierung von mNHOs
Die Synthese einer Reihe neuartiger mNHOs erfolgte durch Deprotonierung von 4-Methyl-1,2,3-triazolium-hexafluor-phosphaten oder -iodiden, die über zwei Syntheserouten erzeugt wurden (Schema 2). Für 1,3-diarylsubstituierte Derivate nutzten wir die bereits für 1 c und 1 f beschriebene9 direkte oxidative (3+2)-Cycloaddition von Triazenen mit Alkinen (Route A).19 Für die 1,3-Dialkyl-substituierten Triazoliumsalze wurde eine schrittweise Synthese über die Methylierung der bisher in der Literatur nicht beschriebenen 1,3-Diisopropyl-substituierten 1,2,3-Triazol-5-ylidene20 gewählt (Route B).21 Die Deprotonierung der Triazoliumsalze mit Kaliumbis(trimethylsilyl)amid (KHMDS) lieferte die entsprechenden mNHOs 1 a–1 h in hohen Ausbeuten. Sowohl elektronenziehende (1 a, 1 b) als auch elektronenschiebende (1 d) und sterisch anspruchsvolle (1 e) Substituenten R′ wurden toleriert.

Synthese Triazol-abgeleiteter mNHOs 1 a–1 h (Dipp=2,6-Diisopropylphenyl, Tripp=2,4,6-Triisopropylphenyl). Unten links: Röntgen-Festkörperstruktur von mNHO 1 g. Die thermischen Ellipsoide (bei 100 K) sind mit einer Wahrscheinlichkeit von 50 % dargestellt. Ausgewählte Bindungslängen (in Å): C1−C2 1.368(2); C2−C3 1.431(2); C3−N1 1.355(2); N1−N2 1.317(2); N2−N3 1.356(2).
Die sterisch sperrigen N-Dipp-gruppen sind nicht zwingend erforderlich, um stabile mNHOs zu erhalten. Deren Synthese kann auch mit N-iPr-Gruppen durchgeführt werden (1 g). Selbst vollständig alkylsubstituierte mNHOs sind synthetisch zugänglich, wie die Isolierung von mNHO 1 h zeigt. Die 13C NMR-Verschiebungen der exocyclischen =CH2-Einheit in 1 a–1 h liegen im Bereich von 48.3 bis 39.6 ppm und sind damit im Vergleich zu regulären terminalen Olefinen erheblich Hochfeld-verschoben. Die Kristallisation von mNHO 1 g ergab Einkristalle, die für die Röntgenbeugung geeignet waren.21, 22 Die Einkristall-strukturanalyse zeigt, dass die C1−C2-Bindungslänge [1.368(2) Å] von 1 g viel länger ist als die von vergleichbaren Olefinen (Schema 2 und Abbildung S32).21 Während herkömmliche NHOs typischerweise farblose Feststoffe sind,4 wurden die mesoionischen NHOs 1 unter inerter Atmosphäre als intensiv gefärbte Verbindungen isoliert. Das Absorptionsmaximum von mNHOs variiert und verschiebt sich bathochrom mit abnehmender Elektronendichte (Abbildung 1A, B). Wie in Abbildung 1C dargestellt, korrelieren die Absorptionsmaxima von mNHOs, λmax, linear mit der Energie des LUMOs, ϵLUMO (Abbildung 1C).21

(A) Optisches Erscheinungsbild der Lösungen von mNHOs 1 a–1 h in THF sowie (B) ihre UV/Vis-Spektren in THF und (C) die lineare Beziehung von λmax mit ϵLUMO.
Wie die schnelle Entfärbung an der Luft zeigte, sind mNHOs äußerst luftempfindliche Verbindungen. Wurden sie jedoch unter einer inerten Atmosphäre gelagert, erwiesen sich die mNHOs 1 a–1 f sowohl in Lösung als auch im festen Zustand tagelang als stabil. Die N-Alkyl-substituierten mNHOs 1 g und 1 h sind zersetzungsanfälliger (z. B. beim Erhitzen auf 50 °C), aber bei Raumtemperatur unter inerter Atmosphäre mindestens 24 h lang stabil.21
Kinetische Experimente: Nucleophilie der mNHOs
Bei der Auswahl der Referenzelektrophile für die Bestimmung der nucleophilen Reaktivitäten der mNHOs verwendeten wir Michael-Akzeptoren, deren Elektrophilie E zuvor zuverlässig bestimmt worden waren.18 Die in Schema 3 dargestellten Produktstudien dienten also hauptsächlich dazu, Michael-Akzeptoren zu finden, mit denen es möglich sein sollte, die Nucleophilieparameter N und sN der mNHOs 1 mit Hilfe der Kinetik der Kohlenstoff-Kohlenstoff-Bindungsbildungsreaktion zwischen mNHO und Elektrophil zu bestimmen. Zunächst untersuchten wir die Reaktionen von N-Aryl- und N-Alkyl-substituierten mNHOs 1 c und 1 g mit p-Chinonmethiden (pQMs) 4 a und 4 b in THF. Die zwitterionischen Produkte 6 a–6 c wurden in 52 bis 88 %iger Ausbeute isoliert und spektroskopisch analysiert. Das pQM/mNHO-Addukt 6 a wurde zusätzlich durch Röntgenbeugung charakterisiert.21, 22 Mehrere Kombinationen zeigten jedoch deutliche Überlagerungen der UV/Vis-Absorptionen von mNHOs und pQMs, was photometrische kinetische Studien nicht erlaubte.

Reaktivität von mNHOs 1 gegenüber elektronenarmen Olefinen 4 oder 5 (Ausbeuten beziehen sich auf isolierte, kristallisierte Produkte).
Die Arylidenmalonate 5 a–5 h sind eine weitere Serie schwach reaktiver Michael-Akzeptoren (E=−17.7 bis −23.8), die zuvor bereits als Referenzelektrophile für die Charakterisierung hoch reaktiver Nucleophile vorgeschlagen wurden.23 Wie in Schema 3 dargestellt, ergaben die Reaktionen von Arylidenmalonaten 5 mit mNHOs 1 c oder 1 g zwitterionische Addukte 7 a–7 c, die mit spektroskopischen Methoden und im Falle von 7 c zusätzlich mit Röntgenkristallographie vollständig charakterisiert wurden.21, 22 Selbst die Reaktion des sterisch gehinderten tBu-substituierten mNHO 1 h mit dem nur schwachen Elektrophil 5 h (E=−23.80) ergab das entsprechende Additionsprodukt 7 d. Da ihre UV/Vis-Absorptionen nur wenig mit denen der mNHOs 1 überlappen, verwendeten wir die Michael-Akzeptoren 5 als Referenzelektrophile, um die Nucleophilie der elektronenreichen mNHOs 1 durch kinetische Studien zu bestimmen.
Unter der Annahme analoger Reaktionswege für alle weiteren mNHO/Arylidenmalonat-Kombinationen wurde die Kinetik der Adduktbildung zwischen den mNHOs 1 und den Referenzelektrophilen 5 in THF (20 °C) photometrisch verfolgt, wobei der Abfall der UV/Vis-Absorptionen einer der beiden farbigen Reaktionspartner bei λmax mit Hilfe von Stopped-Flow-Techniken verfolgt wurde. Unter Bedingungen pseudo-erster Ordnung (einer der Reaktionspartner wurde in mindestens 4-fachem Überschuss eingesetzt) wurden die Geschwindigkeitskonstanten 1. Ordnung kobs (s−1) durch Anpassung (Methode der kleinsten Fehlerquadrate) der Exponentialfunktion At=A0 exp(−kobst)+C an den experimentell beobachteten Abfall der zeitabhängigen Absorptionswerte ermittelt. Für jede Kombination von mNHO 1 mit 5 wurde kobs bei vier verschiedenen Konzentrationen der im Überschuss verwendeten Verbindung bestimmt, was es möglich machte, die Geschwindigkeitskonstanten 2. Ordnung k2 (M−1 s−1) aus den Steigungen der linearen Beziehung von kobs mit der Konzentration der Überschusskomponente zu berechnen. Der Ablauf dieses Verfahrens ist in Abbildung 2 für die Kombination von 5 c und 1 b dargestellt. Einzelheiten zu allen kinetischen Messungen sind in den Hintergrundinformationen zu finden. Die so ermittelten Geschwindigkeitskonstanten 2. Ordnung k2 sind in Tabelle 1 aufgeführt.

(A) Ablauf der kinetischen Messungen am Beispiel der Reaktion von 1 b mit 5 c in THF bei 20 °C. (B) Änderungen des UV/Vis-Absorptionsspektrums während der Reaktion von mNHO 1 b mit Arylidenmalonat 5 c in THF. (C) Monoexponentieller Abfall der Absorption von 1 b bei λmax=578 nm im Verlauf der Reaktion mit 5 c ([5 c]0=27.5 mM). (D) Bestimmung der Geschwindigkeitskonstanten 2. Ordnung k2=5.93 M−1 s−1 aus der Steigung der linearen Korrelation der Geschwindigkeitskonstanten 1. Ordnung kobs (s−1) mit [5 c]0.
Elektrophile |
Elektrophilie E[a] |
k2 (M−1 s−1) |
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|---|---|
|
|
1 a |
1 b |
1 c |
1 d |
1 e |
1 f |
1 g |
1 h |
5 a |
−17.67 |
– |
2.07×102 |
1.15×103 |
2.46×103 |
8.11×101 |
5.16×103 |
– |
– |
5 b |
−18.06 |
2.48×101 |
1.19×102 |
6.73×102 |
1.14×103 |
4.19×101 |
3.30×103 |
– |
– |
5 c |
−20.55 |
– |
5.93 |
2.36×101 |
– |
– |
– |
– |
– |
5 d |
−21.11 |
1.36 |
– |
1.07×101 |
2.03×101 |
6.36×10−1 |
1.11×102 |
1.06×105 |
– |
5 e |
−21.47 |
8.04×10−1 |
2.25 |
5.68 |
– |
3.79×10−1 |
4.60×101 |
5.21×104 |
– |
5 f |
−23.10 |
1.90×10−1 |
4.90×10−1 |
– |
2.02 |
– |
– |
9.39×103 |
– |
5 g |
−23.40 |
– |
– |
– |
– |
– |
– |
4.50×103 |
2.48×104 |
5 h |
−23.80 |
– |
– |
– |
– |
– |
– |
3.38×103 |
1.97×104 |
8[b] |
−24.52 |
– |
– |
– |
– |
– |
– |
– |
6.77×103 |
N (sN) |
21.36 (0.42) |
22.30 (0.49) |
22.80 (0.60) |
23.55 (0.56) |
20.78 (0.61) |
24.86 (0.52) |
30.25 (0.54) |
31.92 (0.52) |
|
- [a] Aus Lit. [18, 23]. [b] Für das hochreaktive mNHO 1 h wurde zur Bestimmung von N und sN auch k2 der Reaktion mit Zimtsäureethylester (8) berücksichtigt.
Korrelationsanalyse
In Abbildung 3 sind die dekadischen Logarithmen der Geschwindigkeitskonstanten 2. Ordnung (lg k2) der Reaktionen der mNHOs 1 a–1 h mit den Elektrophilen 5 (und 8) gegen die zuvor bestimmten Elektrophilieparameter E aufgetragen. Aus den linearen Korrelationen wurden unter Verwendung von Gleichung (1) Nucleophilie N und nucleophil-spezifische Suszeptibilität sN der mNHOs 1 in THF berechnet (Tabelle 1).

Lineare Beziehungen von lg k2 der Reaktionen der mNHOs 1 a–1 h mit den Referenzelektrophilen 5 (und 8) in THF bei 20 °C mit den Elektrophilieparametern E von 5 (und 8); die entsprechende Korrelation für 1 e ist in den Hintergrundinformationen dargestellt.
Die Suszeptibilitäten sN liegen in einem engen Bereich (0.42–0.61), was bedeutet, dass die relativen Reaktivitäten der mNHOs 1 gegenüber den Elektrophilen 5 nur geringfügig von der Elektrophilie des Reaktionspartners abhängen. Die Installation von N-Alkyl- statt N-Aryl-Substituenten am Triazolring erhöht die Reaktivität der mNHOs drastisch: Das mNHO 1 g (N-R=N-iPr) reagiert 10.000-mal schneller als das ansonsten strukturell analoge N-Dipp-Derivat 1 c (Abbildung 3).
Diese Ergebnisse führen wir auf ein Zusammenspiel von sterischen und elektronischen Faktoren zurück. Wie aus Röntgenstrukturanalysen und quantenchemischen Berechnungen hervorgeht,21 sind die sterisch anspruchsvollen N-Dipp-Substituenten orthogonal zur Triazolebene ausgerichtet. Deshalb wirken diese Arylringe als Elektronenakzeptoren und führen dazu, dass mNHOs mit N-Dipp- (1 a–1 f) weniger nucleophil sind als solche mit N-iPr-Substituenten (1 g, 1 h). Die sterische Abschirmung durch den Tripp-Substituenten in 5-Position (1 e) führt zu einer noch geringeren Nucleophilie (N=20.78).
Abbildung 4A zeigt, dass die Geschwindigkeitskonstanten 2. Ordnung (lg k2) für mNHO Reaktionen mit 5 linear mit den Hammett-Parametern σ24 der aromatischen Substituenten an 5 korrelieren. Da auch die Nucleophilie N der 5-arylsubstituierten mNHOs 1 a–1 d gut mit Hammetts σ korreliert (Abbildung 4), ist eine einfache Vorhersage der Reaktivitäten von bisher unbekannten mNHOs mit analogen Strukturen möglich. Eine klassische Hammett-Auftragung von lg k2(5 b) gegen σ für die mNHOs 1 a–1 d ergibt eine lineare Beziehung (r2=0.98) mit einer Steigung von −1.46, die der Hammett-Reaktionskonstante ρ entspricht.

(A) Lineare Korrelationen von lg k2 für die Reaktionen 1+5 in THF bei 20 °C mit den Hammett-Substituentenkonstanten σp von 5. (B) Lineare Beziehung von N (aus Tabelle 1) mit Hammett σ für mNHOs 1 a–1 d.
Der Vergleich der Reaktivitäten der mNHOs 1 a–1 h mit anderen nucleophilen Verbindungen in Abbildung 5 zeigt, dass die N-Alkyl-substituierten mNHOs sogar noch reaktiver sind als Pyridinium-Ylide und schwach stabilisierte Carbanionen, die bisher die reaktivsten Verbindungen auf der Nucleophilie-Skala waren.18 Die Nucleophilie der N-Dipp-Derivate liegt im gleichen Bereich wie die der reaktivsten NHCs IMes und SIMes und übertrifft die der klassischen NHOs oder anderer häufig verwendeter nucleophiler Katalysatoren (in THF) um mehrere Größenordnungen (zum Beispiel: N=16.12 für DBU; 15.90 für DMAP; 13.59 für Ph3P).18 Mit der Quantifizierung der mNHO-Reaktivitäten können nun Reaktionszeiten mit verschiedenen Typen von Elektrophilen oder neuartige mNHO/Elektrophil-Kombinationen für die Synthese vorhergesagt werden.

Einbindung der mNHOs 1 a–1 h in die Nucleophilie-Skala (die N/sN-Werte in Klammern wurden aus Lit. [18] übernommen und beziehen sich auf Reaktionen in THF, wenn nicht anders angegeben).
Reaktionen der mNHOs- mit weiteren Elektrophiltypen
Michael-Akzeptoren. Die in Schema 3 dargestellte Bildung von Addukten aus mNHOs und pQMs 4 a (E=−17.29)18 und 4 b (E=−14.36)18 steht im Einklang mit der Vorhersage schneller Reaktionen durch Gleichung (1). Wie in Abbildung 3 dargestellt, konnten wir sogar die Geschwindigkeitskonstante 2. Ordnung k2(20 °C)=6.77×103 M−1 s−1 der Reaktion 1 h+8 verwenden, um die durch die Datenpunkte gebildete lineare Korrelation für die Kinetik von 1 h mit den Arylidenmalonaten 5 g und 5 h zu erweitern.
Aufgrund ihrer hohen Nucleophilie N sind für alle mNHOs 1 schnelle Reaktionen mit den meisten Elektrophilen in der Mayr-Datenbank der Reaktivitätsparameter zu erwarten, die derzeit Reaktivitäten bis hinunter zu E=−29.6 (für 3-Methylcyclohex-2-en-1-on) abdeckt.18 So wird beispielsweise für die Reaktion von mNHO 1 c mit dem schwachen Elektrophil 8 (E=−24.52) durch Gleichung (1) eine Geschwindigkeitskonstante von 0.1 M−1 s−1 vorhergesagt. Damit übereinstimmend lieferte die Reaktion von 1 c mit Zimtsäureethylester (8) in THF das Additionsprodukt, das zu 9 tautomerisierte (Schema 4A).21 Isolierte Einkristalle von (E)-9 waren für Röntgenbeugung geeignet, die die mesoionische Struktur von 9 bestätigte. Der dadurch verringerte Doppelbindungscharakter der exocyclischen π-Bindung in 9 erklärt das NMR-spektroskopisch beobachtete (E/Z)-Gemisch von 9, das beim Lösen von kristallinem (E)-9 in d6-Benzol erhalten wurde.21, 22

Reaktivität von mNHOs 1 gegenüber Michael-Akzeptoren (A) 8, (B) 10 und (C) 12 (Ausbeuten beziehen sich auf isolierte Produkte).
Das Elektrophil 10 (ca. E=−9 bis −10) reagiert mit sekundären Aminen durch einen Angriff des Amins am Furylring und anschließende Ringöffnung zu Stenhouse-Addukten.25 Im Gegensatz dazu ergab die mNHO-Addition von 1 c an die elektronenarme CC–Doppelbindung von 10 das Addukt 11 (Schema 4B). Auch das Arylidenmalononitril 12 (E=−13.30)18 ging eine klassische Michael-Addition mit mNHO 1 c ein, die glatt und in hoher Ausbeute 13 lieferte (Schema 4C).
Tabelle 2 zeigt, dass die experimentellen Geschwindigkeitskonstanten (k2exp) für die Reaktionen des schwach reaktiven mNHO 1 a mit tert-Butylacrylat (14) und β-Nitrostyrol 15 sowie für die Adduktbildung des hoch reaktiven mNHO 1 h mit Zimtsäurenitril (16) mit den nach Gleichung (1) berechneten Werten innerhalb eines Faktors 6 übereinstimmen. Es kann daher davon ausgegangen werden, dass die in Tabelle 1 angegebenen mNHO-Nucleophilieparameter eine allgemeine Vorhersage der Raten von mNHO-Reaktionen mit gewöhnlichen Michael-Akzeptoren mit einer Genauigkeit von etwa einer Größenordnung ermöglichen.
|
||||
mNHOs |
Elektrophile (E) |
k2exp (M−1 s−1) |
k2Gl 1 (M−1 s−1) |
k2exp/k2Gl 1 |
|---|---|---|---|---|
1 a |
14 (−20.22)[a] |
1.77 |
3.0[b] |
1/1.7 |
1 a |
15 (−14.70)[a] |
3.81×103 |
6.3×102 [b] |
6.1 |
1 h |
16 (−24.60)[a] |
8.82×103 |
6.4×103 [b] |
1.4 |
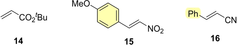
- [a] Elektrophilieparameter E aus Lit. [18]. [b] Geschwindigkeitskonstanten 2. Ordnung k2Gl 1, berechnet durch Einsetzen der Reaktivitätsparameter E, N und sN in Gleichung (1).
Elektrophile Fluorierung. Wir überprüften die Anwendungsbreite von Gleichung (1) für mNHOs weiter, indem wir die Reaktivitätsparameter für mNHO 1 c (N=22.80, sN=0.60) mit dem Reaktivitätsparameter E=−8.44 des elektrophilen Fluorierungsreagenzes NFSI (17) kombinierten.26 Diese Reaktivitätsdaten lassen eine schnelle Bildung (k2≈108 M−1 s−1) des fluormethylsubstituierten Triazoliumsalzes 18 bei Raumtemperatur erwarten. In Übereinstimmung mit dieser Vorhersage und ohne weitere Optimierung lieferte das Mischen von 1 c und 17 in Toluol bei Raumtemperatur innerhalb von 15 min das Salz 18 in einer Ausbeute von 68 % (Schema 5).

Fluorierung von 1 c durch NFSI (17).
CO2 Fixierung. Viele konventionelle NHOs bilden Addukte mit Kohlenstoffdioxid,4d, 27 für das E=−16.3 aufgrund seiner Reaktivität gegenüber dem Indenid-Anion geschätzt wurde.28 Da die mNHOs stärkere Nucleophile als gewöhnliche NHOs sind, erwarteten wir, dass mNHOs ebenfalls Addukte mit CO2 bilden. Diese Erwartung bestätigte sich, da die N-Aryl- und N-Alkyl-substituierten mNHOs 1 c und 1 g mit CO2 in guten Ausbeuten die zwitterionischen Triazoliumcarboxylate 19 a bzw. 19 b lieferten (Schema 6). Die neu gebildete CC-Bindung in 19 a (N-Dipp) erwies sich als schwach. Erhitzen auf 50 °C oder Anlegen eines Vakuums führten zum Verlust von CO2 und setzten das mNHO 1 c frei.21 Daraufhin wurde 19 a unter einem CO2-Strom getrocknet. Unter CO2-Atmosphäre konnten Kristalle erhalten werden, die für die Einkristall-Röntgenanalyse geeignet waren.21, 22 Die Länge der C−CO2-Bindung in 19 a [1.5708(18) Å] ist ähnlich wie die in CO2-Addukten von NHOs [1.549(3)-1.598(6) Å].27

Reaktivität der mNHOs 1 c und 1 g gegenüber CO2. Das reversibel gebildete mNHO-Carboxylat 19 a wurde durch Bestimmung der Einkristall-Röntgenstruktur charakterisiert (Lit. [21, 22]).
Im Gegensatz dazu ist 19 b, das CO2-Addukt von 1 g (N-iPr), bei erhöhten Temperaturen oder im Vakuum stabil. Diese CO2-Bindungsstudie zeigt, dass 1 g (mit N-iPr) eine wesentlich stärkere Lewis-Base ist als 1 c (mit N-Dipp), was mit der Nucleophilie-Reihenfolge (1 g>1c) für diese beiden mNHOs übereinstimmt.
Tolmans elektronische Parameter der mNHOs
Die Gesamt-Donoreigenschaften der mNHO-Liganden 1 c und 1 f wurden zuvor durch Tolmans elektronische Parameter (TEP) charakterisiert, die sich für einen bestimmten Liganden L aus den IR-Carbonylstreckschwingungen in [(L)RhCl(CO)2]-Komplexen errechnen lassen.29 Um die Donoreigenschaften von 1 g mit denen von 1 c9 und 1 f9 zu vergleichen, stellten wir den Rhodiumcarbonylkomplex 20 aus 1 g und Rhodiumcarbonylchlorid her (Schema 7). Nach eindeutiger Charakterisierung der Struktur von 20 durch Röntgenbeugung bestimmten wir die Daten aus den IR-Spektren. In Dichlormethanlösung liegen die IR-Carbonylstreckschwingungen von 20 bei v=2053.1 und 1970.6 cm−1. Der gemittelte Wert vav=2011.9 cm−1 für 20 (entspricht TEP=2029.7 cm−1 berechnet mit TEP=0.8001 vav+420 cm−1 aus Lit.29), liegt deutlich unter den Werten für starke Kohlenstoffdonoren wie NHCs (vav≈2038 cm−1)29 und deutet darauf hin, dass mNHO 1 g ein außergewöhnlich starker Donor-Ligandist. Interessanterweise ist der vav-Wert für 20 fast identisch mit vav der analogen [(mNHO)RhCl(CO)2]-Komplexe von 1 c (vav=2012.5 cm−1) und 1 f (2012.0 cm−1).9 TEPs spiegeln also nicht die stark unterschiedliche Nucleophilie und Lewis-Basizität (siehe unten) der mNHOs 1 c und 1 g wider, was die allgemeine Anwendbarkeit von TEP zur Vorhersage der nucleophilen Eigenschaften von mNHOs einschränkt.

Synthese des Rhodium-Carbonyl-Komplexes 20. Die thermischen Ellipsoide der Röntgen-Festkörperstruktur sind mit einer Wahrscheinlichkeit von 50 % dargestellt (Lit. [21, 22]).
Quantenchemische Berechnungen
Brønsted-Basizität korreliert oft schlecht mit Nucleophilie, insbesondere wenn strukturell unterschiedliche Systeme mit verschiedenen funktionellen Gruppen und Ladungen verglichen werden.30, 31, 32 Alternativ sollten Methylkation-Affinitäten (MCAs), d. h. die negativen Gibbs-Energien ΔG298 der Reaktion eines Nucleophils mit einem Methylkation, die kinetisch beobachteten Bindungsbildungen besser widerspiegeln. Nach einer umfassenden Analyse von Baldi, Van Vranken et al. korrelierten die MCAs von neutralen und negativ geladenen (mehrheitlich C-zentrierten) Nucleophilen gut mit ihren nucleophilen Reaktivitäten (N⋅sN), wenn ein COSMO(∞)-Lösungsmittelmodell in den quantenchemischen Berechnungen verwendet wurde.33
In einem analogen Ansatz berechneten wir die MCAs der mNHOs 1 a–1 h (Abbildung 6A, C)34 mit dem SMD-Kontinuum-Solvatationsmodell für THF auf dem Theorieniveau B3LYP/def2-TZVPP//BLYP/def2-TZVPP. Die Korrelation der Nucleophilie N mit den MCA-Werten der mNHOs 1 a–1 h war im Allgemeinen gut (R2=0.8895). Allerdings war das mNHO 1 e, das den sterisch anspruchsvollen Tripp-Substituenten trägt, ein offensichtlicher Ausreißer (Hintergrundinformationen, S. S146).

(A) Definitionsreaktion für Methylkation-Affinitäten (MCAs), die in dieser Arbeit auf dem Theorieniveau B3LYP(SMD=THF)/def2-TZVPP//BLYP/def2-TZVPP berechnet wurden. (B) Blickrichtung für die Darstellung der “buried volumes“ (%Vbur) an der exocyclischen CH2-Gruppe von mNHOs am Beispiel der topographischen sterischen Karten für 1 a und 1 h. (C) Lineare Beziehung der Nucleophilie N von 1 a–1 h mit einer Linearkombination von Methylkationen-Affinitäten (MCA) und sterischen (%Vbur) Deskriptoren. Die Dreiecke für die Strukturen 21–24 zeigen die Nucleophilie N, die auf Grundlage der dargestellten linearen Korrelationslinie vorhergesagt wurde. (D) Hypothetische mNHO-Moleküle 21–24 mit berechneten MCA- und %Vbur-Werten und vorhergesagter Nucleophilie N.
Daher verwendeten wir die optimierten Strukturen aus den MCA-Berechnungen zur Bestimmung der “buried volumes“ (%Vbur),35 um zusätzlich Variationen des sterischen Anspruchs an der reaktiven exocyclischen Methylengruppe der mNHOs zu berücksichtigen (Abbildung 6B, C).21 Die lineare Kombination beider Deskriptoren, die Beiträge aus den Methylkationen-Affinitäten und der Sterik (d. h. %Vbur) berücksichtigt, beschrieb die experimentellen Nucleophilie N für die gesamte Gruppe der untersuchten mNHOs 1 a–1 h zuverlässig (n=8, R2=0.9726, Abbildung 6C). Auf Grundlage dieser Ergebnisse wird das Design neuartiger mNHOs mit vorhersagbarer Nucleophilie durch ressourceneffiziente DFT-Berechnungen möglich sein, wie am Beispiel der mNHO-Strukturen 21–24 gezeigt wird (Abbildung 6C, D).21
Schlussfolgerungen
Die Persistenz von mesoionischen N-heterocyclischen Olefinen (mNHOs) mit sperrigen 2,6-Diisopropylphenylgruppen an den Stickstoffatomen wurde bereits früher erkannt.9 Wir haben nun beobachtet, dass mNHOs auf Triazolbasis mit einer Vielzahl von Substituenten, einschließlich 1,3,5-trialkylierter Varianten, bei Raumtemperatur persistente Substanzen sind. Die Kinetik der Reaktionen von mNHOs mit Arylidenmalonaten als Referenzelektrophilen zeigte, dass ihre Nucleophilie durch Variation der Substituenten über einen weiten Bereich hinweg (N=20.8 bis 31.9) flexibel eingestellt werden kann. Alkyl-substituierte mNHOs sind deutlich nucleophiler als ihre Aryl-substituierten Analoga und derzeit die stärksten Nucleophile auf der umfassenden Nucleophilie-Skala der Münchener Gruppe, die momentan >1000 C-, N-, O-, P-, S- und Se-Nucleophile sowie fast 200 Hydrid-Donoren umfasst.18 Tolmans elektronische Parameter sind kein Indikator für die außerordentlich hohe Nucleophilie der Alkyl-substituierten mNHOs. Die Nucleophilie-Parameter N der mNHOs korrelieren hingegen gut mit einer linearen Kombination ihrer Methylkation-Affinitäten (d. h. DFT-berechnete Lewis-Basizitäten) und “buried volumes“ %Vbur (d. h. sterische Hinderung am Reaktionszentrum), was die Entwicklung neuartiger mNHO-Strukturen mit vorhersehbarer, maßgeschneiderter Reaktivität und Affinität gegenüber Elektrophilen erleichtert. In dieser Arbeit wurde exemplarisch gezeigt, dass mit den Reaktivitätsparameter N und sN Raten und Produkte der Reaktionen von mNHOs mit verschiedenen elektrophilen Reaktionspartnern (Michael-Akzeptoren, Kohlenstoffdioxid, elektrophile Fluorierungsreagenzien) korrekt vorhergesagt werden können. Wir kommen daher zu dem Schluss, dass diese Parameter eine zuverlässige Grundlage für eine systematische Untersuchung des synthetischen Potenzials dieser neuen Verbindungsklasse bieten.
Danksagung
Wir danken Dr. Julian Holstein (TUD) und Dr. Peter Mayer (LMU) für ihre Hilfe bei der Röntgenstrukturbestimmung. Wir danken der Deutschen Forschungsgemeinschaft (NMR: DFG-Projekt 452669591) im Rahmen des deutschen Exzellenzclusters EXC2033-Projektnummer 390677874-RESOLV und dem Emmy-Noether-Programm (HA 8832/1-1). Finanziert/mitfinanziert von der Europäischen Union (ERC, CC-CHARGED, 101077332). Die Autoren danken dem Leibniz-Rechenzentrum (www.lrz.de) für die zur Verfügung gestellten Rechen- und Datenressourcen. Diese Forschung wurde ganz oder teilweise durch den Österreichischen Wissenschaftsfonds (FWF), Projekt Nr. J-4592 (Erwin Schrödinger-Stipendium an A.E.) gefördert. Zum Zwecke des offenen Zugangs hat der Autor eine CC BY Public Copyright-Lizenz auf jede von ihm akzeptierte Manuskriptversion angewandt, die aus dieser Einreichung hervorgeht. Open Access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.
Interessenkonflikt
Die Autoren erklären, dass keine Interessenkonflikte vorliegen.
Open Research
Data Availability Statement
Die Daten, die die Ergebnisse dieser Studie unterstützen, sind in den Hintergrundinformationen zu diesem Artikel verfügbar.

