

Dynamic Kinetic Resolution of 2-(Quinolin-8-yl)Benzaldehydes: Atroposelective Iridium-Catalyzed Transfer Hydrogenative Allylation
These authors contributed equally to this work.
] co-first authors.
Abstract
An atroposelective Ir-catalyzed dynamic kinetic resolution (DKR) of 2-(quinolin-8-yl)benzaldehydes/1-naphthaldehydes by transfer hydrogenative coupling of allyl acetate is disclosed. The allylation reaction takes place with simultaneous installation of central and axial chirality, reaching high diastereoselectivities and excellent enantiomeric excesses when ortho-cyclometalated iridium-DM-BINAP is used as the catalyst. The racemization of the substrates occurs through a designed transient Lewis acid-base interaction between the quinoline nitrogen atom and the aldehyde carbonyl group.
Axially chiral biaryl scaffolds, particularly those containing N-heterocycles, exist in a number of natural products, biologically active compounds, privileged ligands, and catalysts.1 However, in sharp contrast to the well-established approaches for the asymmetric synthesis of axially chiral biaryls,2 the atroposelective synthesis of chiral (iso)quinoline derivatives remains challenging. Among the main reasons stand their lower configurational stability and their tendency to coordinate metal catalysts.3 Therefore, extensive efforts have been made to synthesize these axially chiral compounds using different designed strategies that circumvent these problems. Among them, CoI- or RhI-catalyzed [2+2+2] cycloadditions between nitriles and alkynes,4 C−H functionalization reactions5 or Pd-catalyzed dynamic kinetic asymmetric transformations of racemic heterobiaryl (pseudo)halides6 are remarkable. However, these approaches have been applied mainly to the synthesis of 1-arylisoquinolines and their derivatives but methods to obtain 8-arylquinoline atropisomers have rarely been reported. These include kinetic resolution by asymmetric transfer hydrogenation,7 atroposelective halogenation using a bifunctional Cinchona-derived organocatalyst,8 Suzuki–Miyaura cross-coupling reaction,9 and enantioselective C−H olefinations using a Pd/chiral phosphoric acid as catalyst.5d, 10 Chiral quinoline derivatives, however, are appealing core structures in many bioactive compounds and natural products.11 Regarding axially chiral derivatives, quinoline BI 224436 I has been identified as the first non-catalytic site integrase HIV inhibitor reaching a clinical trial12 (Figure 1). Additionally, an axially chiral quinoline derivative II has recently been identified as a lead for the development of HIV-1 RNase H inhibitors.13 Finally, axially chiral quinoline derivatives such as III have also found applications as P,N ligands in asymmetric catalysis.14 Collectively, this information reveals that further development of efficient methodologies for the synthesis of axially chiral 8-arylquinolines is highly desirable.

Selected axially chiral quinoline derivatives.
Inspired by our original findings regarding the configurational lability of ortho‘-borylated 2-aryl isoquinolines,15 we recently demonstrated that relatively weak Lewis acid-base interactions (LABIs) between a strategically located acidic functionality (as weak as a carbonyl group) and a functional group containing a basic nitrogen atom can be used to decrease the energy barriers of atropisomerization. In this scenario, any transformation that eliminates the acidic character of the carbonyl group by quaternization consequently increases the racemization barriers, eventually resulting in configurationally stable compounds. This strategy was first exploited in the atroposelective synthesis of chiral heterobiaryl carbinols through an asymmetric Zn-catalyzed hydrosilylation of configurationally labile heterobiaryl ketones.16 In this transformation, racemization through transition state TSI is facilitated by the presence of a five-member structure that alleviates the steric repulsion and/or distortions by widening the angles ϕ1 and ϕ2 (Scheme 1A). More recently, we reported a DKR of N-arylindole 2-carbaldehydes by atroposelective bioreduction.17 In this case, the five-member pyrrole ring in TSII, with ϕ1–ϕ4 angles >120°, is also key to keep the steric repulsion at low levels and locate the formyl and the nitrogen atom at optimal distances to facilitate the N⋅⋅⋅CHO LABI interaction. In contrast, exploiting LABI interactions to resolve prototypical biaryl systems in which the cyclic structure enclosing the bonding interaction and both aryl rings are six-membered is a more challenging task. In fact, the atropisomerization of 1-formyl-2′-(dimethylamino)biphenyls through TSIII proved to be beyond the limit for a reasonable resolution under mild conditions18 due to an unfavorable balance of repulsive/attractive forces in the N(sp3)⋅⋅⋅CHO interaction (Scheme 1B). Motivated by the interest mentioned above in quinoline derivatives, however, we envisioned that the planar geometry of the quinoline in 2-(quinoline-8-yl)benzaldehydes 1 should significantly alleviate the steric repulsive interaction in the transient N(sp2)⋅⋅⋅CHO Lewis pair formed in TSIV.

Racemization based on Lewis acid-base interactions for the atroposelective DKR of heterobiaryls.
To disable the Lewis acid character of the carbonyl and consequently enable a DKR, we decided to focus on the Ir-catalyzed asymmetric carbonyl allylation developed by the Krische group.19 This process, affording quinoline-derived atropisomers 2 bearing central and axial chirality, represents the first example of the Krische allylation for the construction of axial chirality. Furthermore, unlike the previous LA-LB-based DKRs based on carbonyl reduction, a new C−C bond is formed, offering additional synthetic possibilities for the resulting products.
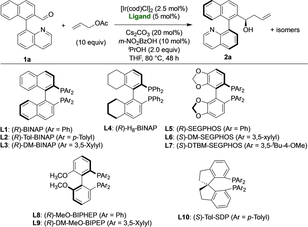
The optimization of the atroposelective C-allylation between 1 a and allyl acetate was then carried out (Table 1). The reaction is catalyzed by a cyclometalated iridium complex generated in situ from [Ir(cod)Cl]2 and C2-symmetric bidentate phosphine ligands. We first evaluated the suitability of various of these ligands, in combination with Cs2CO3 and m- nitrobenzoic acid, with iPrOH as hydrogen donor, in THF at 80 °C. To our delight, (R)-BINAP (L1) provided the desired product in good conversion (85 %), 3.6 : 1 d.r. and high enantioselectivity (95 % ee for both diastereomers, Table 1, entry 1). Better diastereoselectivities were observed for (R)-Tol-BINAP (L2) and (R)-DM-BINAP (L3) (entries 2 and 3), while the enantioselectivities remained high. On the contrary, the use of (R)-SEGPHOS (L5) (entry 5) or (R)-MeO-BIPHEP (L8) (entry 8) led to an erosion of the diastereoselectivity, although full conversion was achieved in both cases. Disappointingly, (R)-H8-BINAP (L4) (entry 4) and (R)-DM-MeO-BIPHEP (L9) (entry 9) provided moderate reactivities, while no reaction resulted using (S)-DTBM-SEGPHOS L7 (entry 7) and (S)-Tol-SDP L10 (entry 10).20 The effect of the solvent was explored next, using the best ligands L2, L3, and L6. With L2, conversions and selectivities in 1,4-dioxane or DME were similar to those in THF (Table 1, entries 11 and 12). On the other hand, lower conversions and d.r.’s were observed using L6 in these two solvents (entries 15 and 16), while MTBE, CH2Cl2 and more polar solvents were shown to be detrimental for the reaction.21 To our delight, better d.r.’s were observed using L3 in combination with both 1,4-dioxane and DME (6.3 : 1 d.r.), while conversions and ee's remained excellent (entries 13 and 14). Finally, the reaction could be completed in 20 h by heating at 100 °C, while maintaining the same d.r. and ee (entry 17). In view of these results, we established the use of 2.5 mol% of [Ir(cod)Cl]2, 5 mol% of (R)-DM-BINAP, 20 mol% of Cs2CO3, 10 mol% of m-nitrobenzoic acid and 2 equiv. of iPrOH in DME at 100 °C, for 20 h, as optimal reaction conditions.22
|
|||||
Entry[a] |
L |
Solvent |
conv. (%)[b] |
dr[b] |
ee (%)[c] major/minor |
|---|---|---|---|---|---|
1 |
L1 |
THF |
85 |
3.6 : 1 |
95/95 |
2 |
L2 |
THF |
97 |
4.2 : 1 |
96/95 |
3 |
L3 |
THF |
89 |
4.6 : 1 |
96/94 |
4 |
L4 |
THF |
74 |
3.2 : 1 |
89/93 |
5 |
L5 |
THF |
>99 |
2.9 : 1 |
98/97 |
6 |
L6 |
THF |
76 |
4.2 : 1 |
98/95 |
7 |
L7 |
THF |
<5 |
n.d. |
n.d./n.d. |
8 |
L8 |
THF |
>99 |
3.0 : 1 |
96/98 |
9 |
L9 |
THF |
75 |
3.8 : 1 |
86 : 85 |
10 |
L10 |
THF |
<5 |
n.d. |
n.d./n.d. |
11 |
L2 |
1,4-Dioxane |
97 |
4.1 : 1 |
96/94 |
12 |
L2 |
DME |
95 |
4.4 : 1 |
96/94 |
13 |
L3 |
1,4-Dioxane |
87 |
6.3 : 1 |
97/96 |
14 |
L3 |
DME |
>99 |
6.3 : 1 |
97/98 |
15 |
L6 |
1,4-Dioxane |
57 |
3.6 : 1 |
96/92 |
16 |
L6 |
DME |
39 |
3.5 : 1 |
96/90 |
17[d] |
L3 |
DME |
>99[e] |
6.3 : 1 |
97/99 |
- [a] Reactions at 0.1 mmol scale. n.d.: Not determined. [b] Determined by 1H NMR spectroscopy. [c] Determined by HPLC on chiral stationary phases. [d] At 100 °C; reaction time 20 h. [e] The major (Ra,R)-2 a isomer was isolated in 75 % yield after column chromatography.
We then explored the scope of the reaction using different configurationally labile heterobiaryl aldehydes 1 a–z23 (Scheme 2). The allylation reaction by DKR was successfully applied to naphthaldehyde derivatives to obtain alcohols 2 a–k, bearing central and axial chirality, in good to high yields and diastereoselectivities and excellent enantioselectivities in all cases. Aldehyde 1 b, with a methyl substituent at position 5 of the quinoline, provided the corresponding allylic alcohols 2 b with good diastereoselectivity (8.3 : 1 d.r.), excellent enantioselectivity (98 % ee) and 84 % yield. Substrates with electron-withdrawing substituents at position 5 of the quinoline (1 c–1 f) were also well tolerated, providing good diastereoselectivities (5.1–6.4 d.r.) and excellent ee’s (94–98 % ee). The yield and d.r. slightly decreased for an electron-donating methoxy group at this position (2 g), while the enantioselectivity remained excellent. Substrates 1 h–1 j bearing electron-withdrawing substituents at position 6 of the quinoline ring were also tolerated, affording good overall yields and diastereoselectivities and excellent enantioselectivities. Furthermore, when a methyl group was placed at this position (1 k), higher diastereoselectivity (7 : 1 d.r.) and a good 82 % yield were observed, while maintaining a high 95 % ee. Quinoline substrates 1 l–z, bearing a 2-formyl-6-methylphenyl residue at position 8 also afforded the desired products 2 l–z in good to high yields and diastereoselectivities and excellent enantioselectivities. Electron-withdrawing substituents at position 5 of the quinoline ring, such as fluoro- (2 n), trifluoromethyl- (2 o) and chloro (2 q) were well tolerated, providing d.r.’s ranging between 4.2 : 1 and 6.7 : 1, and high enantioselectivities (91 %-96 % ee). The highest diastereoselectivity was obtained for product 2 p bearing an ester functionality at position 5 (10 : 1 d.r.), with excellent 96 % ee. The presence of a nitro group at position 5 was also well tolerated, affording moderate yield and diastereoselectivity but excellent enantioselectivity. Furthermore, placing an electron-donating methoxy group at position 4 of the quinoline ring (2 s) resulted in a better diastereoselectivity, (7.5 : 1 d.r.), 50 % yield, with high 94 % ee. Products bearing halides (2 u, 2 v and 2 w) and methyl (2 x) functionalities at position 6 were also well tolerated in terms of yield and selectivity. Finally, ortho-chlorinated derivatives 1 y and 1 z were also found to be suitable substrates for the reaction, affording the corresponding products 2 y and 2 z with similar levels of diastereo- and enantioselectivity. It must be noted that, without exceptions, the diastereomers could be easily separated by flash chromatography. To showcase the practical value of the methodology, a large scale (1.63 mmol) DKR of quinoline aldehyde 1 a was performed under the standard conditions, affording the products 2 a in good 75 % yield, the same excellent enantioselectivity (97 % and 99 % ee for the major and the minor diastereomers, respectively) and improved diastereoselectivity (8 : 1 d.r.).

Atroposelective allylation of 8-[(2′-formyl)aryl]quinolines 1 a–z by DKR. Yields correspond to pure major diastereomers after chromatography. Ee′s were determined by HPLC on chiral stationary phases. [a] In brackets, data for the reaction carried out at 1.63 mmol scale.
Next, we analyzed the suitability of other allyl acetates in the reaction. To this end, crotyl- and 2-methylallyl acetate were tested under the optimized conditions using 1 a as substrate. Unexpectedly, the desired allyl alcohols were not observed, and a cyclic lactam 3 was isolated instead, whose structure was confirmed by X-ray diffraction analysis22 (Scheme 3A). The larger size of the crotyl and 2-methylallyl groups in combination with the steric hindrance of aldehyde 1 a likely prevent carbonyl allylation. Compound 3 is believed to be formed from 1 a through an iridium-catalyzed prototropy. Additional control experiments were then performed to support the formation of transient N⋅⋅⋅CHO Lewis pairs as the racemization pathway. Krische carbonyl allylation can be performed from either the aldehyde or the alcohol oxidation level.19 In this regard, racemic biaryl alcohol 4 a was subjected to the optimized reaction conditions in the absence of iPrOH. After 20 h, product 2 a could be isolated in 75 % overall yield and with similar enantioselectivities for both isomers, although with a slightly decreased 3.5 : 1 d.r. (Scheme 3B). On the other hand, the reaction performed with configurationally stable rac-(1,1′-binaphthalene)-2-carbaldehyde 5 as the substrate afforded the allylation product 6 in 66 % overall yield with 2.5 : 1 d.r. and excellent enantioselectivities for both diastereomers (Scheme 3C). Unreacted (Sa)-5 was also recovered in 30 % yield and 80 % ee,24 showing that a simple kinetic resolution takes place in this case. The same scenario was predicted for isoquinoline substrate 7 (Scheme 3D), but a lack of reactivity was observed instead, which could be explained by deactivation of the catalyst by coordination of the more accessible isoquinoline N atom to the cyclometalated iridium intermediate.

A: Attempts of allylation with other allyl acetates. B: Allylation from alcohol 4 a. C: Kinetic resolution of rac-(1,1′-binaphthalene)-2-carbaldehyde 5. D: Reactivity of isoquinoline derivative 7.
To demonstrate the versatility of the resulting products, heterobiaryl alcohol (Ra,R)-2 a was subjected to a variety of derivatization reactions (Scheme 4). First, butadiene derivative 8 was obtained in good yield through the dehydration of 2 a. Furthermore, biaryldiol (Ra,R)-9 was prepared by a hydroboration-oxidation reaction in moderate yield but without erosion on optical purity. Aminoalcohol derivative (Ra,R)-10 was also prepared by selective catalytic hydrogenation of the quinoline ring using NiCl2 ⋅ 6H2O/NaBH4. Additionally, phosphorus functionalities were introduced by reaction with chlorodiphenylphosphine or (R)-BINOL chlorophosphite, yielding organophosphinate (Ra,R)-1125 or phosphite (Ra,R,Ra)-12, respectively. The absolute configuration of the former was determined by single-crystal X-ray analysis22 and hence, the configuration of the major isomer (Ra,R)-2 a was assigned as the same (Scheme 4). On the other hand, the configuration of the minor diastereomer was assigned by chemical correlation after dehydration of (Sa,R)-2 a and comparison of the HPLC traces with those obtained for (Sa)-8, which afforded the opposite enantiomer (Ra)-8. The absolute configuration of all other products was assigned by analogy.

Transformations from (Ra,R)-2 a.
In summary, we have successfully developed a novel DKR of axially chiral 2-(quinolin-8-yl)benzaldehydes by Ir-catalyzed transfer hydrogenative coupling of allyl acetate. This new methodology features broad scope and good functional-group tolerance, scalability and affords products in good yields and diastereoselectivities with high levels of enantioselectivity. The formation of a transient Lewis pair between the nitrogen atom in the heterocycle and the aldehyde group is responsible for the racemization of the substrates through the formation of 6-membered cyclic transition states. The synthetic versatility of the resulting products has been validated with their transformation into appealing derivatives with potential use as ligands in asymmetric catalysis, without erosion of the optical purity.
Acknowledgments
We thank the Spanish Ministerio de Ciencia e Innovación (Grants PID2019-106358GB-C21, PID2019-106358GB-C22, contracts RYC-2017-22294 for V.H. and BES-2017-081561 for P. R-S), European funding (ERDF), and Junta de Andalucía (Grants P18-FR-3531, P18-FR-644, US-1262867, and US-1260906). We also thank Dr. Francisco José Fernández de Córdova (IIQ-CSIC) for X-ray diffraction analysis.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.

