

Excimer- und Exciplex-Bildung in durch aurophile Wechselwirkungen präkonditionierten Gold(I)- Komplexen
Abstract
Excimere und Exciplexe sind definiert als Aggregate von Atomen oder Molekülen A/A′, in denen es nur in angeregten Zuständen [A2]* und [AA′]* zu interatomaren/-molekularen Bindungen kommt. Ihre Bildung wurde weithin bekannt durch ihre Bedeutung in Gasphasen-Lasertechnologien. Jüngste Untersuchungen in der Goldchemie haben der Excimer- und Exciplex-Chemie ein neues Gebiet eröffnet, das hauptsächlich durch die Aggregation von Gold(I)-Verbindungen (AuI(5d10)) über aurophile Kontakte bedingt wird. In den zugehörigen Excimeren entstehen Au–Au-Bindungen mit Bindungsenergien und -abständen, die denen im Grundzustand von Au0(5d106s1)- und AuII(5d9)-Verbindungen entsprechen. Der aktuelle Kenntnisstand über Excimere und Exciplexe in der Goldchemie wird hier vorgestellt, vorwiegend an einfachen Beispielen wie der Assoziation von [Au(CN)2]−-Anionen in Festkörpern und in Lösung. Die Excimer-Bildung führt zu einem breiten Spektrum an photophysikalischen Effekten, deren Relaxationsdynamik in einigen Fällen kürzlich aufgeklärt wurde. Excimere sind aktuell auch von großer Bedeutung auf dem Gebiet der “photoredox binuclear gold catalysis”.
1 Einführung
Die Stukturchemie von Gold(I) wird im Wesentlichen durch ihre niedrige Koordinationszahl (CN=2) mit zwei in linearer Anordnung fest gebundenen, elektroneutralen (L) oder anionischen (X) Liganden in den drei Kombinationen [L−AuI−L]+, [L−AuI−X] und [X−AuI−X]− charakterisiert (A–C
 ). Obwohl diese Anordnung noch offenen Zugang zu den zentralen Goldatomen bietet, sind diese Metallzentren schlechte Elektrophile, und nur die stärksten weichen Donoren werden unter Bildung von Komplexen mit CN>2 aufgenommen. Die für die Unterbringung eines zusätzlichen Liganden erforderliche Abwinkelung der L/X-AuI-L/X-Achsen ist mit einem erheblichen Energieverlust verbunden, der über die Bildungsenergie einer dritten oder vierten AuI-L/X-Bindung nur schwer zu kompensieren ist.1-5 Die Vorliebe der Gold(I)-Zentren für CN=2 liegt an starken relativistischen Effekten, die nicht nur zu einer Kontraktion der anionischen/kationischen Radien führen, sondern auch die Orbitalcharakteristika des Kations modifizieren und für die Bindung Hybride mit hohem s-Charakter favorisieren.6-9
). Obwohl diese Anordnung noch offenen Zugang zu den zentralen Goldatomen bietet, sind diese Metallzentren schlechte Elektrophile, und nur die stärksten weichen Donoren werden unter Bildung von Komplexen mit CN>2 aufgenommen. Die für die Unterbringung eines zusätzlichen Liganden erforderliche Abwinkelung der L/X-AuI-L/X-Achsen ist mit einem erheblichen Energieverlust verbunden, der über die Bildungsenergie einer dritten oder vierten AuI-L/X-Bindung nur schwer zu kompensieren ist.1-5 Die Vorliebe der Gold(I)-Zentren für CN=2 liegt an starken relativistischen Effekten, die nicht nur zu einer Kontraktion der anionischen/kationischen Radien führen, sondern auch die Orbitalcharakteristika des Kations modifizieren und für die Bindung Hybride mit hohem s-Charakter favorisieren.6-9
Umgekehrt ermöglicht die lineare Zweifachkoordination von Gold(I) eine enge gegenseitige Annäherung seiner Komplexe unter Ausbildung kurzer AuI⋅⋅⋅AuI-Kontakte (gezeigt in D und E, mit n=2− bis 2+). Selbst mit sperrigen Liganden L/X sind aufgrund der freien Drehbarkeit der Molekülachsen unter Ausbildung jeder zwischen gestaffelt und ekliptisch möglichen Konformation enge Au⋅⋅⋅Au-Kontakte realisierbar. Bemerkenswerterweise wird die Linearität der L/X-Au-L/X-Achsen durch diese Au-Au-Kontakte nicht stark beeinflusst. In zwei- oder mehrkernigen Gold(I)-Komplexen können diese Au⋅⋅⋅Au-Kontakte durch verbrückende, bifunktionelle Donor-Liganden unterstützt werden, was zu offenkettigen oder cyclischen Anordnungen führt, deren geometrische Details von der Starrheit der verbindenden Liganden bestimmt wird. Es werden konformative Änderungen induziert, die einen Ringschluss über den Au⋅⋅⋅Au-Kontakt ermöglichen (F) oder die transannularen Au⋅⋅⋅Au-Wechselwirkungen verstärken (G
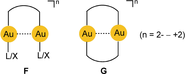 ).
).
Die Annahme einer durch diese Kontakte zwischen zwei neutralen Molekülen [L−Au−X] hervorgerufenen signifikanten kovalenten Au-Au-Bindung ist deshalb nicht sofort eingängig, weil die AuI-Kationen eine Elektronenkonfiguration mit abgeschlossener Schale (5d10) aufweisen und wegen der Polarisierung der L-Au- und Au-X-Bindungen dieselbe elektrische Ladung (<+1) tragen, die eher zu einer Coulomb-Abstoßung führen sollte; es bleiben folglich nur Dispersionskräfte mit sehr geringen Bindungsenergien als mögliche Quelle. Trotzdem gibt es eine Fülle experimenteller Belege, und auch die Ergebnisse umfangreicher quantenchemischer Berechnungen haben gezeigt, dass diese AuI⋅⋅⋅AuI-Wechselwirkungen tatsächlich mit Energiebeiträgen einhergehen, die über Standard Van-der-Waals-Wechselwirkungen hinausgehen und vergleichbar sind mit anderen schwachen Wechselwirkungen wie der Wasserstoffbrückenbindung.10-13 Für Gold wurde das Phänomen Aurophilie genannt, was eine direkte attraktive Wechselwirkung zwischen Au-Atomen impliziert,14 aber inzwischen wurde erkannt, dass der Terminus zur Einbeziehung der meisten Nachbarelemente im Periodensystem, insbesondere der schweren, bei denen relativistische Effekte die Elektronenkonfiguration stark beeinflussen, auf Metallophilie erweitert werden sollte.15
Die Aggregation von [X−Au−X]−-Anionen oder [L−Au−L]+-Kationen zu oligomeren Anionen {[X−Au−X]−}n oder Kationen {[L−Au−L]+}n ist sogar noch bemerkenswerter, da es sich hier um den besonderen Fall einer anti-elektrostatischen Assoziation handelt, d. h. sie erfolgt entgegen der Coulomb-Abstoßung von Ionen. In dieser Hinsicht erinnert sie an die kürzlich für die Anionen HCO3−, HSO4− und H2PO4− zusammengefasste anti-elektrostatische Wasserstoffbrückenbindung.16 Diese Parallele zeigt auch die vergleichbaren Effekte dieser beiden schwachen Kräfte, Wasserstoffbrückenbindung und Aurophilie. Drei spezifische Beispiele für anti-elektrostatische Aurophilie sind unten gezeigt (Dicyanido- und Dithiocyanatoaurat(I)-Anionen sowie Di(ammin)gold(I)-Kationen).
Die Tatsache, dass intramolekulare aurophile Wechselwirkungen maßgeblich zur strukturellen Organisation zwei- oder mehrkerniger Gold(I)-Verbindungen und auch zur Aggregation und Packung von ein- und mehrkernigen Gold(I)-Verbindungen in Kristallen und in Lösung beitragen können, hat viele Konsequenzen für die Eigenschaften der verschiedenen anorganischen und metallorganischen Systeme. In den letzten Jahren wurde in umfangreichen Forschungsaktivitäten insbesondere das photochemische Verhalten von Gold(I)-basierten Materialien im festen Zustand untersucht.17 Diese Arbeiten haben früh erkennen lassen, dass die Emissionseigenschaften vom Au-Au-Abstand in den Kristallen abhängen.18 Die derzeit untersuchten Phänomene schließen viele unterschiedliche Gebiete ein, wie die Thermo-, Mechano-, Solvato- und Vapochromie unter Standard- oder extremen Drücken, und es wurde gefunden, dass Gold(I)-Verbindungen mit intra- oder intermolekularen Au⋅⋅⋅Au-Kontakten alle möglichen Arten von durch mehrere Stimuli ausgelösten lichtemittierenden Eigenschaften zeigen.19-22
Die chemische Reaktivität von Gold(I)-Verbindungen wird ebenfalls von aurophilen Wechselwirkungen beeinflusst, da diese zu Anordnungen von Komplexen führen, welche direkte Zwei- oder Mehrzentren-Reaktionswege ermöglichen. Schema 1 zeigt Beispiele für eine transannulare Addition an zweikernige Gold(I)-Komplexe (d10–d10) unter Bildung von lange gesuchten Gold(II)-Komplexen (d9–d9) oder von Produkten mit gemischten Oxidationsstufen (X, Y=Halogen, X2=Disulfid RSSR).23-25

Oxidative Addition an cyclischen und offenkettigen zweikernigen Gold(I)-Komplexen.
Für einkernige Gold(I)-Komplexe sind ähnliche Phänomene in Lösung selten, da die schwachen aurophilen Wechselwirkungen durch effiziente Solvatisierung der einkernigen Einheiten leicht aufgehoben werden können. Bei hohen Konzentrationen der Komplexe und bei niedrigen Temperaturen erhöht sich die Zahl der Aurophilie-basierten Dimere oder Oligomere allerdings, und die Assoziation kann über verschiedene analytische Techniken nachgewiesen werden, insbesondere durch Verfolgung der UV/Vis-Absorptions- und -Emissionseigenschaften.26 In allen Diskussionen muss daran erinnert werden, dass Au⋅⋅⋅Au-Wechselwirkungen – als schwache Kräfte – fast immer nur kleine Beiträge zur Gesamtenergie eines gegebenen Systems liefern, und dass nur eine sorgfältige Evaluierung aller Parameter ein konsistentes Bild des tatsächlichen Beitrags vergleichbarer chemischer und physikalischer Phänomene ergibt. Jüngste Veröffentlichungen beziehen sich auf diese inhärenten Probleme,27, 28 auf die auch in den meisten früheren Aufsätzen zu diesem Thema hingewiesen worden ist.11-14
Erwartungsgemäß sind intermolekulare aurophile Wechselwirkungen sogar noch ausgeprägter für Aggregate aus zwei- und mehrkernigen Gold(I)-Komplexen, bei denen mehr als ein Au-Au-Kontakt ausgebildet werden kann. Frühere wie auch Beispiele aus jüngster Zeit spiegeln die sich mehrenden Anzeichen für diese Wechselwirkungen wider (H
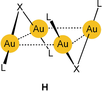 : X=Cl, Br; L=PR3; Gesamtladung 2+).29, 30
: X=Cl, Br; L=PR3; Gesamtladung 2+).29, 30
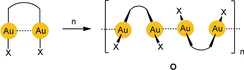
In diesem Aufsatz dirigieren die Autoren den Leser in Richtung des spezifischen Phänomens der [Au⋅⋅⋅Au]-Excimer- und -Exciplex-Bildung, die durch eine Aurophilie-basierte Präorganisation der Komponenten stark gefördert wird. In diesen Excimeren [Au−Au]*, bei denen es sich um angeregte Zustände der aurophilen Aggregate handelt, sind die beiden Goldatome fester aneinander gebunden als in den Aggregaten der Vorstufen im Grundzustand (Schema 2; n=0 oder 2). Zur Markierung der Excimer-Bindung, auf die sich dieser Bericht fokussiert, wurden in den Formeln hier und weiter unten rote Linien eingeführt.

Photochemische Erzeugung von Excimeren aus ein- und zweikernigen Gold(I)-Komplexen.
Überraschenderweise wurde dieses Phänomen in experimentellen und theoretischen Untersuchungen nur sporadisch aufgegriffen, und die Ergebnisse – obwohl frühe Beispiele schon vor etwa 25 Jahren publiziert worden waren – haben kaum Beachtung gefunden. Die [Au−Au]*-Bindung in den Excimeren repräsentiert eine interessante Zwischenstufe zwischen den starken Au0-Au0- und AuII-AuII-Bindungen auf der einen, und den schwachen aurophilen AuI⋅⋅⋅AuI-Wechselwirkungen auf der anderen Seite. Impulsgeber für die jüngsten Arbeiten waren frühe Beobachtungen und Untersuchungen der Gruppen von Fackler, Che und Anderen, aber die meisten langfristigen Weiterentwicklungen stammen von den Gruppen von Patterson und Omary, ergänzt durch jüngere theoretische und experimentelle Untersuchungen von Yam, Iwamura, Kim, Thiel, Yersin und verschiedenen Anderen, wie unten gezeigt wird.
Um die Grundlagen für diese Präsentation zu schaffen, werden im ersten Teil die etablierten Fälle einer “echten” Au-Au-Einfachbindung mit hohem kovalenten Charakter (5d106s1–5d106s1 für Au0, und 5d9–5d9 für AuII) eingeführt, die die nötigen Referenzdaten für Bindungslängen und -energien und deren Variation in Abhängigkeit externer Parameter liefern. Daran anschließend folgt eine kurze Zusammenfassung der entsprechenden Daten für inter- und intramolekulare aurophile Wechselwirkungen, welche nicht nur von der Wahl der Liganden X und L und deren Substitutionsmuster abhängen, sondern eben auch von externen Einflüssen. Im letzten und aktuellsten Kapitel werden dann die derzeit verfügbaren Daten über Gold(I)-Excimere und -Exciplexe in diesem Gesamtkontext vorgestellt und diskutiert.
Da ähnliche Phänomene in Verbindungen von AuIII(d8) nicht auftreten, wird dieser höhere Oxidationszustand nicht in die Betrachtungen mit einbezogen. Die enorme Komplexität der photophysikalischen Eigenschaften von Gold-Clustern und -Nanopartikeln – mit einem durchschnittlichen Oxidationszustand der Goldatome im Cluster zwischen Au0 und AuI – sprengt ebenfalls den Rahmen dieses Aufsatzes, in dem versucht wird, die Excimer-/Exciplex-Effekte anhand einfacher Beispiele aufzuzeigen.
2 Eckdaten für Gold-Gold-Bindungen in zweikernigen Au0- und AuII-Verbindungen
2.1 Die Gold-Gold-Bindung im Au2-Molekül und seinen Komplexen
Zweiatomige Goldmoleküle Au2 sind Bestandteile von Golddampf, der durch Erhitzen von metallischem Gold (Siedepunkt 3080 K) in Öfen auf über 2000 °C oder durch Laserablation und -verdampfung erzeugt wird. Spektroskopische Untersuchungen des hantelförmigen Au2-Moleküls in der Gasphase haben eine lange Geschichte. Die Molekülabmessungen wurden über verschiedene Methoden bestimmt, dazu gehören die resonante Zweiphotonen-Ionisation und supersonische Molekülstrahluntersuchungen. Aus rotationsaufgelösten Spektren ergab sich schließlich ein Au-Au-Abstand von 2.4715 Å im Grundzustand und eine Dissoziationsenergie von 53.0 kcal mol−1.31-35
Dieser Abstand ist erstaunlich empfindlich gegenüber dem Einfluss von Liganden, was über resonanzverstärkte FIR-Multiphotonen-Dissoziationsspektroskopie und quantenchemische Berechnungen gezeigt wurde. Das gilt selbst für die 1:1- und 1:2-Addukte mit Edelgasen, z. B. für die kürzlich untersuchten Addukte Au2⋅Kr und Au2⋅Kr2 (Schema 3, L=Kr). In diesen linearen Molekülen (C∞v, D∞h Symmetrie) wurde eine geringfügige Verkürzung der Au-Au-Abstände von 2.429 (für Au2) auf 2.421 und 2.40 Å (für Au2⋅Kr bzw. Au2⋅Kr2) berechnet (MP2-Rechnungen).36-38

Das Molekül Au2 und seine Komplexe.
Versuche, stabile Au2-Komplexe zu erhalten, waren lange erfolglos,39, 40 bis 2013 der Gruppe von Bertrand die Synthese eines Bis(carben)-Komplexes gelang.41 Durch Einsatz seiner effizienten CAAC-Liganden wurden stabile kristalline (CAAC)Au−Au(CAAC)-Komplexe erhalten, deren Molekülstrukturen ermittelt werden konnten (Schema 3, rechts: L=CAAC). Überraschenderweise wurde ein Au-Au-Abstand von 2.5520 Å gefunden. Die in dieser Arbeit durchgeführten quantenchemischen Berechnungen ergaben Au-Au-Abstände für das freie Au2-Molekül und seinen Komplex von 2.546 bzw. 2.579 Å, was auf eine leichte Schwächung der Au-Au-Bindung bei der Komplexbildung hindeutet. Für den bisher noch nicht zugänglichen Phosphin-Komplex (H3P)Au−Au(PH3) wurde ein Abstand von 2.550 Å vorhergesagt und ein Abstand von 2.562 Å für das Au2-Molekül (MP2).40 Für Au2 gibt es offensichtlich einen deutlichen Unterschied zwischen diesen berechneten und dem experimentellen Wert von 2.4715 Å (siehe oben). In älteren42, 43 und jüngeren37, 38 theoretischen Untersuchungen von Au2 werden Daten um die 2.50 Å (MP2) und wieder solche am kürzeren Ende (MP2: 2.429 Å) erhalten.37, 42-44 Diese Diskrepanz ist momentan noch unerklärt.
Das Au2 Molekül wurde kürzlich als Modellsystem für sogenannte Regium-Bindungen an Goldclustern herangezogen. In diesen Untersuchungen werden die Oberflächen von Aun-Nanopartikeln analysiert, um Bereiche mit einer Präferenz für Donor- und Akzeptor-Wechselwirkungen mit Substratmolekülen zu lokalisieren.45 Das zweiatomige Molekül ist der kleinste “Cluster” und es wurde gefunden, dass es elektrophile σ-Löcher für die Anlagerung von Nukleophilen wie Ammoniak oder Wasser44-46 in Verlängerung der Molekülachse anbietet, welche, was den Abstand betrifft, den aktiven Positionen auf einer reinen Goldoberfläche ähnlich sind.
2.2 Die Gold-Gold-Bindung in zweikernigen Gold(II)-Komplexen
AuII-AuII-Bindungen wurden erstmals über eine transannulare Addition von Oxidationsmitteln an Dimetallacyclen mit zwei Gold(I)-Zentren erzeugt. Das Ligandengerüst bestand aus dreiatomigen Phosphonium-bis-methyliden47, 48 oder Diphosphinomethanen49, 50 mit verbrückenden C-P-C- bzw. P-C-P-Einheiten (G und Schema 1). In späteren Arbeiten kamen auch P-C-S-Linker51 oder Kombinationen mit Dithiocarbamaten52, 53 zum Einsatz. Des Weiteren wurden für eine Vielzahl mono- und polycyclischer Varianten P-C-C-Verbrückungen mit Arylphosphinen genutzt.54, 55 Die jüngsten Variationen verwendeten Metallacyclen mit auf Amidinaten und Guanidinaten basierenden N-C-N-Klammern (Schema 4).56-58 Schließlich wurden auch bifunktionelle Carbene als Liganden in cyclische zweikernige Komplexe eingeführt, allerdings mit größeren Ringen.59 Ein Sonderfall wurde von der Gruppe von Wickleder vorgestellt, der das Gold(II)-Sulfat AuSO4 hergestellt hat. Kristalle dieser Verbindung enthalten einen Achtring und zwei die Gold(II)-Zentren verbrückende Sulfate.60 Die Goldatome werden durch Sulfat-Sauerstoffatome benachbarter Moleküle koordinativ weiter abgesättigt.

Transannulare Au-Au-Bindung in zweikernigen cyclischen Gold(II)-Komplexen.
Wie bereits in Schema 1 gezeigt, wurden als Oxidationsmittel die molekularen Halogene Cl2, Br2, I2 und einige ihrer Interhalogen-Verbindungen, Alkylhalogenide oder Disulfide (RSSR) eingesetzt. Bei der oxidativen Addition bleiben Größe und Form der Metallacyclen weitgehend unverändert, aber die ursprünglich linear zweifach koordinierten Gold(I)-Zentren werden zu Gold(II)-Zentren mit einer quadratisch-planaren Koordinationsumgebung, und der transannulare Au-Au-Abstand verringert sich deutlich (siehe unten). Die neu gebildete Au-Au-Bindung wird also von zwei Ligandenklammern doppelt gehalten. In jüngsten Untersuchungen wurden cyclische AuII-AuII-gebundene Verbindungen auch auf elektrochemischem Weg erhalten.61
Verbindungen mit nicht von Liganden gehaltenen AuII-AuII-Bindungen wurden viel später und oft unerwartet gefunden, z. B. in AuI/AuIII-Komproportionierungs-Reaktionen. Frühe Beispiele wurden von den Gruppen von Yam in 199662, 63 und Kuz′mina in 200364 dokumentiert, gefolgt von denen von Raubenheimer,65 Mathur66 und Bochmann.67, 68 Theoretische Arbeiten von Pyykkö haben die thermodynamische Stabilität der nicht gestützten AuII-AuII-Bindung als einzige Verknüpfung zwischen zwei Moleküleinheiten bestätigt.69 Beispiele sind in Schema 5 gezeigt.

Nicht durch Liganden unterstützte Au-Au-Bindungen in zweikernigen Gold(II)-Komplexen.
Die Au-Au-Abstände in den diauracyclischen Verbindungen (Schema 4) liegen in einem engen Bereich, mit einem unteren Limit von 2.47 und 2.49 Å für die Guanidinate bzw. das Sulfat mit kleinen Stickstoff- bzw. Sauerstoff-Donoratomen57, 60 und einer oberen Grenze von 2.61 Å für die Liganden mit größeren Schwefel- oder Selen-Donoratomen (2.61 bzw. 2.72 Å),51, 52 oder in makrocyclischen Systemen mit ungünstigen Konformationen.59
In ungestützten Beispielen (Schema 5) ist der Bereich ähnlich und überraschend eng zwischen 2.49 und 2.61 Å für Komplexe mit einem C^N^C-Pincer-67 bzw. einem 1,8-Diphosphinonaphthalin-Liganden.62 DFT-Rechnungen von Xiong und Pyykkö haben für zweikernige Modellverbindungen, die den in experimentellen Arbeiten verwendeten Molekülen66 ähneln, AuII-AuII-Abstände von 2.50–2.55 Å und eine Bindungsenergie von ca. 47.8 kcal mol−1 vorhergesagt.69
3 Relevante Aspekte der aurophilen AuI-AuI-Bindung
3.1 Hintergrund
Die Prototypen der aurophilen Bindung wurden in der Einleitung vorgestellt (A–F).1-70 In allen Fällen werden die Goldatome auf Sub-van-der-Waals-Abstände aneinander geführt, sobald die sterischen Voraussetzungen des Systems eine derartige Annäherung erlauben. Diese Anziehung führt in Kationen des Typs {E[Au(L)]n}m+ auch zur spontanen, anti-intuitiven Anhäufung komplexer Gold(I)-Einheiten [(L)Au]+ an einem gegebenen Hauptgruppenelement- oder Übergangsmetall-Koordinationszentrum E.71-75
Als ein erstes Kriterium für eine Charakterisierung wurden Molekül- und Kristallstrukturen analysiert, um einen relevanten Bereich für die Au-Au-Bindung zu erhalten. Überraschenderweise ist in Gold(I)-Komplexen das untere Ende dieser Abstände mit ca. 2.75 Å bereits kürzer als die Summe von zwei kovalenten Radien (2.88 Å in fcc-Goldmetall) und nicht viel länger als die Werte in diamagnetischen zweikernigen Gold(0)- (5d106s1) und Gold(II)-Komplexen (5d9), beide mit einer kovalenten Au-Au-Bindung (2.47–2.61 Å; siehe oben). Am oberen Ende wird um 3.50 Å immer noch eine deutliche Anziehung beobachtet (und berechnet), was nahe an der Summe der Van-der-Waals-Radien von zwei Gold(I)-Atomen (ca. 3.60 Å) liegt.
Ein weiteres strukturelles Kriterium ist die unspezifische Ausrichtung der Bindung, angedeutet durch die Flexibilität der in die Au-Au-Kontakte involvierten relevanten Bindungs- und Torsionswinkel. Zum Dritten kann ein Gold(I)-Zentrum mehr als einen Au⋅⋅⋅Au-Kontakt unterhalten und dadurch Teil oligomerer oder polymerer Aggregate werden, in denen Monomere über aurophile Wechselwirkungen mit oder ohne die Unterstützung von Liganden miteinander verknüpft werden.
All diese strukturellen Beobachtungen wurden durch quantenchemische Berechnungen auf verschiedenen Niveaus bestätigt, welche vor allem eine Abschätzung der Wechselwirkungsenergie als dem wichtigsten Kriterium zum Ziel hatten. Berechnete Energieprofile für die Annäherung zweier einkerniger Modellmoleküle L−Au−X zeigen schwache, aber deutliche Minima für Au-Au-Abstände im oben erwähnten Bereich und sind weitgehend unabhängig von Torsionsbeschränkungen wie einer parallelen (ekliptischen) oder senkrechten (gestaffelten, “gekreuzte Schwerter”) Konformation, wobei letztere nur unwesentlich gegenüber ersterer bevorzugt ist (D, E). Die Wechselwirkungsenergien hängen stark vom Charakter der Liganden ab, können aber bis zu 15 kcal mol−1 erreichen.76-84
Diese theoretischen Ergebnisse stehen in Einklang mit den wenigen experimentellen Daten, die in einer Reihe von temperaturabhängigen NMR-Untersuchungen erhalten wurden und alle im Bereich zwischen 5 und 15 kcal mol−1 liegen.85-87 In theoretischen Berechnungen hat die dimere Modellverbindung {HAu(PH3)}2 einen Au-Au-Abstand von 3.0 Å und eine Bindungsenergie von 13.0 kcal mol−1.88
Dieses Energiekriterium besagt, dass die aurophile Bindung im Bereich der schwachen Kräfte in der Chemie angesiedelt ist, am besten vergleichbar mit Wasserstoffbrückenbindungen, wo die Wechselwirkungsenergien in einem ähnlichen Bereich liegen. Wie bereits erwähnt, zeigen sowohl die Wasserstoffbrückenbindungen als auch die aurophilen Wechselwirkungen eine ähnliche strukturelle Flexibilität in Bezug auf die Kontaktabstände und Winkel. Es gibt auch Freiheitsgrade bezüglich der Anzahl der Kontaktpartner, was für die Aurophilie bedeutet, dass das Referenz-Goldatom mehrere Au⋅⋅⋅Au-Kontakte in größeren Aggregaten ausbilden kann (siehe oben).
3.2 Intermolekulare aurophile Kontakte zwischen einkernigen Gold(I)-Komplexen
Während es für den festen Zustand eine Vielzahl von Beispielen für eine Aggregation von einkernigen Gold(I)-Komplexen über aurophile Wechselwirkungen gibt (siehe oben), ist die Situation für den gelösten Zustand eine andere, wo molekulare oder ionische Einheiten A–C von Lösungsmittelmolekülen umgeben sind und die Solvatationsenergie den Energiegewinn durch aurophile Kontakte übersteigt. Aus diesem Grund findet man – belegt durch Standardmethoden zur Bestimmung von Molekülmassen (Kryoskopie, Ebullioskopie und weitere) oder durch massenspektrometrische Untersuchungen – in verdünnten Lösungen elektroneutraler Moleküle B in unpolaren oder schwach polaren Lösungsmitteln keine Aggregate wie B2- oder Bn-Oligomere.
Mit Entfernen des Lösungsmittels wird die Organisation der elektroneutralen Komponenten B in Kristallen jedoch von aurophilen Wechselwirkungen mitbestimmt, die stärker als normale Van-der-Waals-Kräfte richtungsdirigierend für die Aggregation sind. Die Zusammenlagerung der ionischen Gegenstücke A und C wird weitgehend von Coulomb-Kräften bestimmt, wobei alternierende Ladungen +−+−+−(A⋅⋅⋅C⋅⋅⋅A⋅⋅⋅C⋅⋅⋅A⋅⋅⋅C) bevorzugt sind. Andere Wechselwirkungen wie π-π-Wechselwirkungen oder starke Wasserstoffbrückenbindungen können die Aggregationsform ebenfalls beeinflussen und Au⋅⋅⋅Au-Kontakte ausschließen. Andererseits gibt es viele Fälle, in denen aurophile Wechselwirkungen und Wasserstoffbrückenbindungen einander unterstützend zu zwei- oder mehrkernigen Aggregaten führen.89, 90 Im Folgenden wird das aurophile Verhalten von einkernigen Gold(I)-Komplexen an drei Beispielen gezeigt, die im gegenwärtigen Kontext, nämlich der Bildung von Excimeren, am relevantesten sind.
3.2.1 Der Fall des Dicyanidoaurat(I)-Anions
Ein wichtiges und bestens untersuchtes Fallbeispiel ist die (anti-elektrostatische16) Aggregation des Dicyanidoaurat(I)-Anions [NC−Au−CN]− in Kristallen und Lösungen von Salzen des Typs M+[(Au(CN)2]−, M2+{[Au(CN)2]2}2− und M3+{[Au(CN)2]3}3−. In Kristallen der Alkalimetall-Salze (M=Na, K, Rb, Cs) sind die Anionen in Schichten gepackt, wobei alle Goldatome in einer Ebene liegen und die Cyanid-Gruppen nahezu senkrecht aus dieser Ebene nach oben und unten herausstehen. Das Aggregationsmuster hängt von der Größe der Gegenionen ab, und die Au⋅⋅⋅Au-Kontakte variieren in einem engen Bereich mit einen Durchschnittswert von 3.33 Å.91-96 Abbildung 1 zeigt eine idealisierte {[Au(CN)2]n}n-Schicht mit der am häufigsten gefundenen, hexagonalen Anordnung der Goldatome. In Kristallen von Rb[Au(CN)2], einem Beispiel für eine verzerrte Variante, liegen die Goldatome ebenfalls in einer Ebene, bilden aber vier- und achtgliedrige Ringe.96

Die idealisierte Struktur einer polyanionischen {[Au(CN)2]n}n−-Schicht in Kristallen der meisten Alkali- und Erdalkali-Dicyanidoaurate(I).
Schon in vielen frühen strukturellen Untersuchungen wurde vermutet, dass es “in diesen Festkörpern signifikante Bindungswechselwirkungen zwischen den Goldatomen geben müsse”.97 Derartige niedrig-dimensionale Au⋅⋅⋅Au-Wechselwirkungen wurden auch für das ungewöhnliche Absorptions- und Emissionsverhalten von Alkali-dicyanidoauraten(I) unter hohen Drücken (20 kbar) verantwortlich gemacht. Es wurden extreme Rotverschiebungen bei der Emission beobachtet, die zu den größten für Festkörperverbindungen beobachteten gehören.98 Für kristalline Pulver von K[Au(CN)2] wurden auch in 197Au-Mössbauer-spektroskopischen Untersuchungen bei tiefen Temperaturen (4 K) und hohen Drücken (80 kbar) Anomalien beobachtet, die eine Präferenz für Au⋅⋅⋅Au-Kontakte in der Packung nahegelegt haben.99
In richtungsweisenden Arbeiten wurden auch Salze mit raumfüllenden Kationen wie in NBu4[Au(CN)2], untersucht, welche die Anionen in den Kristallen voneinander trennen,100 sodass die photophysikalischen Eigenschaften isolierter [Au(CN)2]−-Anionen untersucht werden konnten, genauso wie in mit Spuren von M[Au(CN)2] dotierten Kristallmatrizes.101-103 In Kristallen von Salzen mit sehr sperrigen Kationen wie Bis(triphenylphosphoranyliden)ammonium, [(Ph3P)2N]+, kommt es ebenfalls zu keiner Aggregation der Anionen.104
Frühe schwingungsspektroskopische Untersuchungen von Dicyanidoauraten(I) konnten keinen spezifischen Einfluss der Au⋅⋅⋅Au-Kontakte auf die grundlegenden Eigenschaften der Anionen im festen Zustand nachweisen, und IR- sowie 13C-NMR-Spektren (mit 13C-markiertem Cyanid) in Lösung bei verschiedenen Konzentrationen (in Wasser und Dichlormethan bei Raumtemperatur) ergaben keine Anzeichen für größere Effekte aufgrund von Anionen-Aggregation.105
Wie bereits erwähnt, stehen seit einigen Jahrzehnten die Lumineszenz-Eigenschaften von Alkali-dicyanidoauraten(I) im Fokus photophysikalischer Untersuchungen.106, 107 Die meisten dieser Arbeiten haben zu Vergleichszwecken auch die analogen Silberverbindungen mit einbezogen.107 Diese Untersuchungen haben zur Vorstellung von [Au−Au]*-Excimeren und Exciplexen und einem detaillierten Studium ihrer Eigenschaften geführt. Verschiedene quantenchemische Berechnungen der letzten Zeit haben die frühen experimentellen Untersuchungen nachbereitet und das Bild der elektronischen Grund- und angeregten Zustände, insbesondere der Dimere und Trimere (I, J
 ), wie unten dargestellt, verfeinert.108 Die Strukturdynamik dieser Spezies wurde schließlich mittels zeitaufgelöster Pico-/Femtosekunden-Emissions- und -Absorptionsspektroskopie109, 110 und in Lösung über Femtosekunden-Röntgenstreuung111-113 verfolgt.
), wie unten dargestellt, verfeinert.108 Die Strukturdynamik dieser Spezies wurde schließlich mittels zeitaufgelöster Pico-/Femtosekunden-Emissions- und -Absorptionsspektroskopie109, 110 und in Lösung über Femtosekunden-Röntgenstreuung111-113 verfolgt.
Untersuchungen an wässrigen Lösungen oder in unpolaren Lösungsmitteln haben gezeigt, dass darin der Oligomerisierungsgrad mit der [Au(CN)2]−-Konzentration schrittweise zunimmt, was in separaten Konzentrationsbereichen eine Untersuchung von Dimeren, Trimeren und Tetrameren (z. B. 30 mm für das Dimer, 300 mm für das Trimer in Wasser) ermöglicht und eine Verfolgung ihrer Bildung und Umlagerung zulässt. Diese Assoziation erklärt die Beobachtung, dass die Spektren dieser Lösungen deutlich vom Beer'schen Gesetz abweichen. Absorptions- und Lumineszenz-Anregungsspektren von wässrigen oder methanolischen Lösungen zeigen zwischen Raumtemperatur und 77 K (in gefrorenen Gläsern) auch eine Temperaturabhängigkeit. Erwartungsgemäß wird bei tiefer Temperatur ein Anstieg der Aggregation der Anionen zu Oligomeren gefunden. Aus diesen Spektren konnte sowohl die Bildungskonstante als auch die freie Bildungsenergie für das Dimere berechnet werden: K=17.9 m−1; ΔG=−1.86 kcal mol−1 (295 K, 1 atm). Extended-Hückel-Rechnungen mit relativistischen Parametern kamen für das Dimer (ekliptisch/ gestaffelt) zu Au⋅⋅⋅Au-Bindungsenergien von 3.04/ 6.87 kcal mol−1 und Au-Au-Abständen von 3.48/ 2.88 Å.101, 107 Weitere Lumineszenz-Untersuchungen, die viel empfindlicher als Absorptionsmessungen sind, haben gezeigt, dass selbst bei so niedrigen Konzentrationen wie 10−5 m bereits Oligomerisierung eintritt (beobachtet für Methanol-Gläser bei 77 K). Für das Dimer in gestaffelter Anordnung ergaben MP2 Rechnungen einen Au-Au-Abstand von 2.960 Å und eine ν(Au⋅⋅⋅Au)-Streckschwingungsfrequenz von 89.8 cm−1.107 Später lieferten MP2-Berechnungen an Modell-Dimeren Au-Au-Abstände von 3.014 und 2.998 Å für das gestaffelte Dimer bzw. Trimer.108
Die Aggregation von Dicyanidoaurat(I)-Anionen wurde kürzlich auch in ionischen Flüssigkeiten beobachtet. Zeitaufgelöste Lumineszenzspektren zeigten sowohl in Imidazolium- als auch in Pyrrolidinium-Salzen die Anwesenheit verschiedener Oligomere.114
Enthält das Gegenion des Dicyanidoaurat(I)-Anions ein zweifach koordiniertes Gold(I)-Atom – beispielsweise den sperrigen PTA-Liganden (1,3,5-Triaza-7-phosphaadamantan) in [(PTA)Au(PTA)]+ – dann folgt die Aggregation der Sequenz +−+−+− mit alternierenden aurophilen Kontakten zwischen Kationen und Anionen, und es werden keine [Au(CN)2−]n-Oligomere gebildet. Die schlanke, stabförmige Struktur der Anionen passt zur Ausbildung kurzer Au⋅⋅⋅Au-Kontakte (3.271 Å)115 sogar in den reduzierten Raum zwischen den sperrigen PTA-Liganden.
In diesem Zusammenhang hat das Design von Systemen, in denen bestimmte oligomere [Au(CN)2−]2-Einheiten in supramolekularen Matrizes eingeschlossen sind, weitere Möglichkeiten zur Unterscheidung der spezifischen Eigenschaften der Dicyanidoaurat(I)-Oligomere ermöglicht. Mit 24-Pyrimidinium-Krone-6- und 16-Pyrimidinium-Krone-4-Polykationen befinden sich die Dimere, Trimere, Tetramere sowie kettenförmige Aggregate mit vollständig gestaffelter Konformation in den Hohlräumen der kristallinen, supramolekularen Einheiten.117 Kürzlich wurde ein Beispiel beschrieben, bei dem in der Reaktion des Viologens 1,1′-Bis(2,4-dinitrophenyl)-4-4′-bipyridinium-Dichlorid (DNP)Cl2 mit K[AuCN)2] in Wasser ein kristallines Tetrahydrat (DNP)[Au(CN)2]2⋅4 H2O gebildet wird. Das Produkt enthält zweikernige {[Au(CN)2]2}2−-Einheiten mit einer ekliptischen (parallelen) Konformation und einem Au⋅⋅⋅Au-Kontakt am oberen Ende der aurophilen Wechselwirkungen (D, d(Au⋅⋅⋅Au)=3.5108 Å), in Übereinstimmung mit dem für diese Konformation berechneten Wert, die allerdings nicht den Grundzustand in Abwesenheit einer Matrix darstellt.101, 107 Während (DNP)Cl2 und [Au(CN)2]− im sichtbaren Bereich nicht emittieren, zeigt (DNP)[Au(CN)2]2 zwei unabhängige Emissionsbanden in den Regionen der DPN- und [Au(CN)2−]2-Einheiten, wobei es sich bei der ersten um eine durch den interionischen Kontakt zwischen DPN und den Schwermetallatomen hervorgerufene Phosphoreszenz handelt.118
Polymere [Au(CN)2−]n-Stränge können durch Polyammonium-Gerüste wie Polyallylamin-Hydrochlorid-Polymere gestützt werden, welche in Wasser [Au(CN)2]−-Ionen gegen Cl− austauschen. Die Aggregate zeigen eine einstellbare Lumineszenz in Abhängigkeit der relativen Konzentrationen der Komponenten. Bei geringer Anionenzufuhr können die Ketten unter Bildung kleinerer Oligomere aufgebrochen werden. Dieser Prozess kann über die konzentrationsabhängigen Veränderungen in den Absorptions- und Emissionsspektren verfolgt werden.119 Die Verwendung eines Polyrotaxan-Wirts für die [Au(CN)2]−-Anionen liefert kristalline Produkte, in denen das Anion zu symmetrischen Trimeren (J) mit Au-Au-Abständen von 3.103 Å und einem Au-Au-Au-Winkel von 180° assoziiert ist.120 Dieses Ergebnis stimmt auch gut mit berechneten Werten überein.101, 197 Im Gegensatz dazu wurden in der kristallinen Matrix eines Viologen-Akzeptors wie Aza[5]helicen wieder [Au(CN)2−]2-Dimere mit ekliptischer Konformation (d(Au⋅⋅⋅Au)=3.310 Å) gefunden.121 Alle diese Beispiele zeigen den wichtigen Einfluss unabhängiger Kationen auf der einen und strukturierter kationischer Matrizes auf der anderen Seite auf die resultierende Form der Anion-Aggregation.
Die strukturellen und elektronischen Eigenschaften kristalliner Dicyanidoaurate(I) werden stark durch Thallium- oder Platin-Kationen beeinflusst, die wie in Tl[Au(CN)2]122, 123 und [PtII(NH3)4][Au(CN)2]2124 heterometallophile Kontakte ausbilden. Im Gegensatz dazu enthalten die Ketten der [Au(CN)2]−-Anionen im NiII-Komplex [Ni(NH3)2][Au(CN)2]2 keine Nickelatome. Die Unterschiede gehen wiederum auf relativistische Effekte zurück, die z. B. Pt stark von seinem leichteren Homologen Ni abheben.125
Größere {[Au(CN)2]−}n-Aggregate treten in Kombination mit Metallen der ersten Übergangsreihe auf, wo die endständigen N-Atome der Cyanid-Einheiten als Donor-Zentren für eine Vielzahl von Elementen wie Co, Rh, Cu, Cd, In und andere fungieren. In dieser Strukturfamilie liegen häufig kettenförmige [Au(CN)2]−}n-Oligomere und Polymere mit gewinkelt oder zickzackförmig angeordneten Goldatomen vor.126-131 Die Flexibilität der Au⋅⋅⋅Au-Kontakte in Kristallen dieser Verbindungen gestatten eine große räumliche Flexibilität, die sich besonders klar in anomalen (“kolossalen” oder “gigantischen”) positiven und negativen thermischen Ausdehnungen zeigt.132, 133 Frühe Strukturbestimmungen an Dicyanidoauraten(I) der Seltenerd-Elemente lieferten ebenfalls Schichtstrukturen mit in Kagomé-Netzen (drei- und sechsgliedrige Ringe) angeordneten Goldatomen und Au-Au-Abständen von 3.316 Å. Überraschenderweise wurden stark gewinkelte NC−Au−CN-Anionen gefunden, was sowohl kurze Au⋅⋅⋅Au-Kontakte als auch eine optimierte prismatische Koordination der Kationen gestattet.134 Eine anderen Reihe von Untersuchungen befasste sich erneut mit den Strukturen und Lumineszenz-Eigenschaften von Seltenerd Dicyanido-auraten(I), wobei Beispiele mit und ohne aurophile Kontakte gefunden wurden. In Kristallen erfolgt der Energietransfer im ersteren Fall eindeutig über die Au⋅⋅⋅Au-Kontakte.135 Die Untersuchungen wurden auch auf wässrige Lösungen ausgedehnt.136
Bei der Betrachtung der Oligomerisierung von Dicyanidoaurat(I)-Anionen muss unbedingt auch deren Rolle in der technischen Gold-Laugerei und -Aufarbeitung im CIP (carbon-in-pulp)-Prozess erwähnt werden.137 Seit mehr als einem Jahrhundert weiß man, dass Cyanidoaurate(I) aus wässriger Lösung stark an der Oberfläche von Aktivkohle adsorbiert werden, von wo sie bei erhöhten Temperaturen desorbiert und durch alkalische Cyanidlösungen zurückgewonnen werden können. Die beladene Kohle, die zwischen 200 und 20 000 Gramm Gold pro Tonne enthalten kann, ist ein Beispiel für eine sehr dichte Aggregation von Anionen auf Oberflächen, vermutlich in Form von Dimeren.138, 139 Es ist zu beachten, dass andere Gold(I)- oder Gold(III)-Komplexsalze keine vergleichbare Affinität zu Kohlenstoff-Oberflächen zeigen. Offenbar bietet die lineare, fünfatomige Koordinationsgeometrie nicht nur freien Zugang für aurophile Kontakte, sondern auch optimale Bedingungen für eine Adsorption an geeigneten Oberflächen.
3.2.2 Der Fall des Di(thiocyanato)aurat(I)-Anions
In diesem Anion sind die Thiocyanat-Liganden über ihr Schwefelatom an das Gold(I)-Zentrum gebunden, und das komplexe Anion [Au(SCN)2]− hat eine gewinkelte Struktur mit einem Winkel an beiden Schwefelatomen. In grundlegenden Arbeiten auf diesem Gebiet wurde in röntgenstrukturanalytischen Untersuchungen an Kristallen mit Alkali- oder Onium-Kationen gefunden, dass die Anionen {[Au(SCN)2]−}2-Dimere mit aurophilen Kontakten bilden. Die beiden C2-symmetrischen Anionen sind in gestaffelter Konformation assoziiert, und die Verbindungslinie der beiden Goldatome fällt mit der zweizähligen Achse zusammen (K
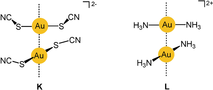 ). Die Au-Au-Abstände variieren mit der Natur des Gegenions, liegen aber alle im engen Bereich von 3.00–3.24 Å. Interessanterweise gilt dies auch für das [NnBu4]+-Salz, wo der Au-Au-Abstand im Dimer 3.07 Å beträgt. Lediglich im [NMe4]+-Salz sind die Anionen in einer gewinkelten Struktur zu Trimeren aggregiert (die Au⋅⋅⋅Au⋅⋅⋅Au-Achse ist am zentralen Goldatom abgewinkelt). Die Maxima in den Absorptions- und Emissionsspektren der Verbindungen (Feststoffe bei 77 K) variieren in Abhängigkeit des Kations, und die Emissionsenergie korreliert mit dem Au-Au-Abstand.140, 141
). Die Au-Au-Abstände variieren mit der Natur des Gegenions, liegen aber alle im engen Bereich von 3.00–3.24 Å. Interessanterweise gilt dies auch für das [NnBu4]+-Salz, wo der Au-Au-Abstand im Dimer 3.07 Å beträgt. Lediglich im [NMe4]+-Salz sind die Anionen in einer gewinkelten Struktur zu Trimeren aggregiert (die Au⋅⋅⋅Au⋅⋅⋅Au-Achse ist am zentralen Goldatom abgewinkelt). Die Maxima in den Absorptions- und Emissionsspektren der Verbindungen (Feststoffe bei 77 K) variieren in Abhängigkeit des Kations, und die Emissionsenergie korreliert mit dem Au-Au-Abstand.140, 141
Die Aggregationsform der [Au(SCN)2]−-Anionen ist ein wichtiges Beispiel für die Stärke der aurophilen Wechselwirkungen. Das SCN−-Anion besitzt ein hartes (N) und ein weiches (S) Donor-Zentrum, welche die Zusammenlagerung der Anionen über N→Au- oder S→Au-Donor-Bindungen unterstützen könnten, aber diese Alternativen werden nicht beobachtet. Stattdessen werden zusätzliche Au⋅⋅⋅Au-Kontakte zwischen den Dimeren gefunden, die zu einer Verlängerung der Anordnungen zu Ketten mit einer Sequenz sich abwechselnder Au⋅⋅⋅Au⋅⋅⋅⋅⋅Au⋅⋅⋅Au⋅⋅⋅⋅⋅Au-Abstände führen.
Zur Klärung der bemerkenswerten Struktur-Lumineszenz-Beziehung wurde in einer anschließenden ausgedehnten Studie das photophysikalische Verhalten der Bis(thiocyanato)aurate(I) untersucht.142 Diese Arbeiten haben zur Identifizierung der einschlägigen Gruppe von Excimeren K geführt (siehe unten).
3.2.3 Der Fall der Diammingold(I)-Kationen [Au(NH3)2]+
Um ein kationisches Gegenstück zum Dicyanidoaurat(I) zu erwähnen, wird kurz das Aggregationsverhalten des Diammingold(I)-Kations (L) in seinen Salzen vorgestellt. Die Kristallstrukturen des Chlorids (als Ammoniat), Bromids, Nitrats und Perchlorats wurden bestimmt, und es wurde übereinstimmend beobachtet, dass die Kationen zu Ketten assoziiert sind, mit Au⋅⋅⋅Au-Kontakten von 3.176, 3.414, 3.091 und 2.990 Å. Die Änderung des Abstands in Abhängigkeit von der Natur des Anions liegt vermutlich an unterstützenden Beiträgen von Wasserstoffbrückenbindungen in der Packung sowohl im Ammoniat des Chlorids wie auch im Nitrat und Perchlorat. Kristalle des Perchlorats zeigen eine starke, breite Emission (450–650 nm), die Metall-Metall-Charge-Transfer(MMCT)-Übergängen zugeordnet wird.143-145
Diese drei Fälle spiegeln sowohl die lange Geschichte als auch die gegenwärtige Relevanz der intermolekularen Aurophilie wider. In jüngsten Arbeiten ist die Variabilität der Liganden L und X für Gold(I) unter Einbeziehung auch immer exotischerer Vertreter zur Bestimmung der homo- und heteroleptischen Verteilung und der Länge und Stärke der aurophilen Kontakte rasch weiter angewachsen.146 Aus Platzmangel kann hier keine umfassende Zusammenfassung geliefert werden.
3.3 Intramolekulare aurophile Kontakte in zwei- und mehrkernigen Gold(I)-Komplexen
Die Zuordnung intramolekularer aurophiler Wechselwirkungen zwischen zwei oder einer größeren Anzahl von Gold(I)-Zentren in einem Molekül (F, G) nur auf der Basis des AuI-AuI-Abstands ist oftmals schwieriger als für die intermolekularen Fälle, da starre Ligand-Geometrien bestimmte Konfigurationen erzwingen können, in denen die Metallzentren in enger Nachbarschaft gehalten werden, ohne dass dies überwiegend durch aurophile Kontakte gelenkt wird.53, 147-149 Bei Liganden mit flexibler Geometrie kann die in einem Kristall gefundene Konformation F mit einem Au⋅⋅⋅Au-Kontakt einfach nur durch Packungskräfte bestimmt werden. Bei Strukturen des Typs G ist eine Ringgröße von mindestens 7 Atomen nötig, damit es zu einer spannungsfreien Anordnung von zwei nahezu linear koordinierten Goldatomen kommen kann. Für Ringgrößen mit 7–9 Atomen sind alle transannularen Au⋅⋅⋅Au-Kontakte kürzer als 3.5 Å. Deshalb werden für eine Charakterisierung der Au-Au-Bindung in Molekülen z. B. des Typs F und G zusätzlich zu Einkristall-Röntgenbeugungsuntersuchungen noch andere analytischen Techniken benötigt.
Frühe Untersuchungen haben sich deshalb auf die Molekulardynamik konzentriert. Die Schwingungsfrequenz-Charakteristika intramolekularer aurophiler Kontakte wurden erstmals für einen zweikernigen Gold(I)-Komplex mit zwei verbrückenden, deprotonierten Ylid-Liganden (M
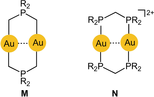 ) studiert, und die Ergebnisse wurden mit den Daten des gleichen Dimetallacyclus von Gold(II) mit einer transannularen, im Verlauf der oxidativen Halogen-Addition (X=Cl, Br, I) (Schema 1) gebildeten AuII-AuII-Bindung verglichen. Es wurde gefunden, dass der Wert für ν(Au⋅⋅⋅Au) von 64 cm−1 im Zuge der Oxidation auf 162, 132 und 103 cm−1 zunimmt, was bestätigt, dass die aurophile Wechselwirkung relativ schwach ist.150 Ähnliche Werte wurden auch für zweikernige Gold(I)-Dimetallacyclen mit Diphosphinomethan-Liganden (N) gefunden, und es wurde eine Korrelation zwischen intramolekularen Au-Au-Abständen und den Kraftkonstanten F(Au⋅⋅⋅Au) gefunden. Es wurden daneben auch Strukturparameter von Grund- und angeregten Zuständen berechnet (siehe unten).151-153 Für die intermolekulare aurophile Wechselwirkung zwischen einkernigen Gold(I)- Komplexen stehen nur Daten aus quantenchemischen Berechnungen zur Verfügung, welche die für die zweikernigen Komplexe bestätigen.
) studiert, und die Ergebnisse wurden mit den Daten des gleichen Dimetallacyclus von Gold(II) mit einer transannularen, im Verlauf der oxidativen Halogen-Addition (X=Cl, Br, I) (Schema 1) gebildeten AuII-AuII-Bindung verglichen. Es wurde gefunden, dass der Wert für ν(Au⋅⋅⋅Au) von 64 cm−1 im Zuge der Oxidation auf 162, 132 und 103 cm−1 zunimmt, was bestätigt, dass die aurophile Wechselwirkung relativ schwach ist.150 Ähnliche Werte wurden auch für zweikernige Gold(I)-Dimetallacyclen mit Diphosphinomethan-Liganden (N) gefunden, und es wurde eine Korrelation zwischen intramolekularen Au-Au-Abständen und den Kraftkonstanten F(Au⋅⋅⋅Au) gefunden. Es wurden daneben auch Strukturparameter von Grund- und angeregten Zuständen berechnet (siehe unten).151-153 Für die intermolekulare aurophile Wechselwirkung zwischen einkernigen Gold(I)- Komplexen stehen nur Daten aus quantenchemischen Berechnungen zur Verfügung, welche die für die zweikernigen Komplexe bestätigen.
In einem Bericht jüngeren Datums wird der Einsatz von EXAFS-Untersuchungen (EXAFS=extended X-ray-absorption fine structure) in Lösung an Molekülen mit einem flexiblen Liganden beschrieben, die belegen sollten, dass die Konformation F in Lösung bestehen bleibt. Für eine ausgewählte Gruppe von Verbindungen mit Bis(phosphin)-Liganden wurde wie erwartet eine Lösungsmittel-abhängige Präferenz für die Aurophilie-stabilisierten Konformeren bestätigt. Die Au-Au-Abstände in zweikernigen Ligand-verbrückten Komplexen stimmen gut mit denen im festen Zustand gefundenen überein, allerding hängen die Abstände sowohl vom Gegenion als auch vom Lösungsmittel ab. Die transannularen Au-Au-Abstände in [(dppm)2Au2]X2 betragen für X=Cl 2.93 Å im Festkörper und 3.08 Å in einer Chloroform-Lösung, für X=BF4 hingegen liegen sie bei 2.90 Å im Festkörper und 2.97 Å in Acetonitril, d. h. das Lösungsmittel bewirkt jeweils eine Verlängerung und Schwächung der aurophilen Wechselwirkung.154
Moleküle mit intramolekularen aurophilen Kontakten (F) können auch mit intermolekular Au⋅⋅⋅Au-verknüpften (O
 ) Oligomeren im Gleichgewicht stehen, je nach Lösungsmittel und pH-Wert mit darin potentiell deprotonierten Liganden.155
) Oligomeren im Gleichgewicht stehen, je nach Lösungsmittel und pH-Wert mit darin potentiell deprotonierten Liganden.155
Es gibt nur sehr wenige Untersuchungen zur intramolekularen Bindung in der Gasphase. In einer kürzlich erschienenen Photoelektronen-spektroskopischen Studie zur Bindung im Au2I3−-Anion wurde über den besonderen Fall einer “Bindungsbiegungs-Isomerie” berichtet.
Für das V-förmige, fünfatomige Anion mit C2v-Symmetrie steht ein Isomer mit überwiegend kovalenter Bindung, einem stumpfen Au-I-Au-Winkel von 100.7° und einem langen Au⋅⋅⋅Au-Kontakt von 3.99 Å mit einem Isomer mit einem spitzen Winkel von nur 72° und einem kurzen aurophilen Au⋅⋅⋅Au-Kontakt von 3.08 Å im Gleichgewicht, das nur um 0.425 kcal mol−1 energiereicher, also praktisch energetisch entartet ist (Schema 6). Bei tiefen Temperaturen wird nur das stumpfwinklige Isomer beobachtet. Dieses Anion ist ein Beispiel für ein Molekül, welches aufgrund zweier konkurrierender chemischer Kräfte zwischen verschiedenen Strukturen oszilliert, was auch durch quantenchemische Berechnungen gestützt wird.156 Größeren Anionen [(AuCl)nCl]− wurden Zickzack-Strukturen mit einer zentralen Kette aus Goldatomen zugewiesen, ähnlich der, die in Kristallen von AuCl gefunden werden.157

Bindungsbeugungs-Isomerie des Anions Au2I3-.
Bei den Untersuchungen der transannularen Au⋅⋅⋅Au-Wechselwirkungen richtete sich das Hauptaugenmerk auf die UV/Vis-Absorptions- und -Emissionseigenschaften.158-160 Typische Beispiele waren Komplexe mit 1,3-difunktionellen Phosphinen, die achtgliedrige Ringe enthielten, in denen zwei nahezu parallele P-Au-P-Achsen eng beieinander gehalten werden (N). Diese Art der Anordnung war Gegenstand einer großen Anzahl experimenteller und theoretischer Untersuchungen.153, 161, 162 Bereits in einer frühen Studie wurden den Grund- und angeregten Zuständen Elektronenkonfigurationen zugewiesen, die eine Interpretation der verschiedenen in Lösung beobachteten Übergänge gestatteten. Es wurde ein stark reduzierender angeregter Zustand mit einem Redox-Potential von −1.6 V gefunden. Ähnliche Resultate wurden auch für dreikernige Analoga erhalten.163 Transannulare aurophile Kontakte wurden auch in strukturanalytischen, spektroskopischen und theoretischen Studien an zwei- und dreikernigen Gold(I)-Komplexen mit einer breitgefächerten Auswahl an Diphosphin-Donoren mit Xanthengerüst untersucht, in jüngster Zeit auch in Verbindung mit Dicyanido-aurat(I)-Anionen.19, 20, 164, 165
Die Arbeiten auf diesem Gebiet wurden später, wie unten genauer beschrieben, durch experimentelle und quantenchemische Untersuchungen ähnlicher cyclischer kationischer Komplexe mit P,S-Donorliganden ergänzt.166 Sogar noch engere Au⋅⋅⋅Au-Kontakte werden mit N,N-difunktionellen Donorliganden ausgebildet, kürzlich auch in zwei- und vierkernigen Guanidinat-Komplexen.167 In ähnlicher Weise wurden vor kurzem auch Phosphin- mit Carben-Donoren (NHCs) kombiniert, um zu Komplexen mit einem achtgliedrigen Ring oder zwei fusionierten achtgliedrigen Ringen mit einem bzw. zwei transannularen Au⋅⋅⋅Au-Kontakten zu gelangen.168 All diese Beispiele und einige neu hinzugekommene Typen wurden, wie in den folgenden Abschnitten zusammengefasst, als Grundzustände für die Bildung von entsprechenden Excimeren herangezogen.
4 Die Bildung und Reaktivität von und die Bindung in [AuI−AuI]*-Excimeren und -Exciplexen
4.1 Hintergrund
Vor etwa 100 Jahren wurde für Quecksilberatome im Gaszustand erstmals die Bildung spektroskopisch beobachtbarer, zweiatomiger Einheiten aus elektronisch angeregten Atomen beobachtet, und kurz darauf auch für Edelgasatome, die bis dahin als notorisch inerte Spezies bekannt waren.169, 170 Zwanzig Jahre später erregte die Bildung langlebiger, angeregter Dimere von Pyren und anderen aromatischen organischen Verbindungen im festen Zustand und in Lösung das Interesse vieler Wissenschaftler.171, 172 Der Terminus Excimer wurde geprägt173 zur Unterscheidung solcher durch Kollision gebildeter angeregter Dimere von kurzlebigen, schwach assoziierten Dimeren, die in relativ hochkonzentrierten Lösungen oder im kristallinen Zustand vorkommen. Die Excimer-Bildung kann experimentell mittels UV/Vis-Absorption, Lumineszenz-Emission, zeitaufgelöster Absorption und Emission sowie Resonanz-Raman-Spektroskopie beobachtet und untersucht werden. Die zeitaufgelöste Röntgenstreuung wurde kürzlich zu einer weiteren Option mit außergewöhnlichen neuen Perspektiven.
Schließlich wurde gefunden, dass auch strukturell privilegierte Komplexe von Übergansmetallen, insbesondere solche mit RhI, Pt0, PtII und AuI, eine große Tendenz zur Bildung von Excimeren zeigen.174 Die Monomere unterliegen im Grundzustand einer schwachen, jetzt als metallophiler Kontakt bezeichneten Anziehung, die die Bildung viel stärkerer kovalenter Metall-Metall-Bindungen im angeregten Zustand vermittelt. In Gegenwart anderer geeignet reaktiver, im Grundzustand befindlicher Spezies führen Wechselwirkungen mit den Excimeren zu Exciplexen. Oligomere Exciplexe, einige davon entstanden aus im Grundzustand befindlichen, schwach assoziierten Dimeren oder Oligomeren, sind ebenfalls bekannt,175 was zu einer steigenden Anzahl von komplexen Excimeren und Exciplexen führte.7 Weitere Untersuchungen des angeregten Zustands wurden durch spezielle photochemische und photophysikalische Eigenschaften wie Thermochromie, optische Speichermöglichkeit und Abstimmbarkeit stimuliert.
Es scheint also, dass die durch Überlappung von Atomorbitalen zweier Goldatome entstehenden kovalenten Bindungen nicht auf Au0 und AuII begrenzt sind (siehe oben): Zweifach koordinierte AuI-Atome können ebenfalls, aber ausschließlich im angeregten Zustand, kovalent gebunden werden. Die Präorganisation durch aurophile Wechselwirkungen [Au⋅⋅⋅Au] (der bekannteste Fall der Metallophilie) im Grundzustand assistiert bei der Bildung der chemischen AuI-AuI-Bindung im entsprechenden Excimer [AuI−AuI]* während der Anregung.
Gemäß einer Metall-zentrierten (MC) LCAO-Näherung sind Metall-Metall-Wechselwirkungen zwischen Übergangsmetallen mit geschlossener d10-d10-Schale repulsiv. Nun tendieren aber wie schon erwähnt metallophile, dispersive Wechselwirkungen dazu, zwei Goldatome mit dieser Konfiguration zusammenzuhalten. Solche Wechselwirkungen können theoretisch nur über die Einbeziehung von Orbitalen angeregter Zustände erklärt werden, wie sie die Methoden der theoretischen Chemie anbieten.7 Theoretisch erscheint die Möglichkeit einer Stärkung der d10-d10-Wechselwirkungen durch photochemische Aktivierung also als logische Konsequenz.
Die Idee, dass die Ausbildung kovalenter Bindungen in geschlossenschaligen d8-d8- und d10-d10-Systemen über elektronische Anregung realisiert werden kann, ist nicht neu. Sie wurde von Gray und Mitarbeitern schon in den 1970er und frühen 1980er Jahren für RhI- und PtII-Komplexe vorgeschlagen. Metallatome, die einen Satz verschiedener einzähniger Liganden tragen, lagern sich in konzentrierten Lösungen paarweise über Metall-Metall-Anziehung eng zusammen,176, 177 wobei sie meist von doppelt verbrückenden Diphosphin-Liganden in enger Nachbarschaft zueinander gehalten werden.178 Experimentelle Hinweise für eine erhebliche RhI−RhI-Wechselwirkung im angeregten Zustand wurden über Resonanz-Raman-Spektroskopie in Lösung erhalten.177
4.2 Excimer-Bindung in zweikernigen AuI-Komplexen mit verbrückenden Phosphin-Liganden
4.2.1 Frühe experimentelle Untersuchungen und Interpretationen
Inspiriert von den Pionierarbeiten der Gray'schen Schule eröffneten die von Ludwig,179 Caspar180 und wiederum Gray181 geführten Untersuchungen der angeregten Zustände von doppelt dppm-verbrückten Komplexen des nullwertigen Palladiums, Platins und Golds neue Forschungsfelder. Über Ergebnisse wurde zwischen 1989 und 1993 in kurzer Abfolge berichtet, so etwa über die spektroskopischen Eigenschaften von Komplexen des Typs [Au2(P^P)2]2+ (P^P=Bis(dimethylphosphino)methan, dmpm; Bis(dimethylphosphino)ethan, dmpe; Bis(diphenylphosphino)methan, dppm), [Au2(P^P)3]2+ (P^P=dmpm) und [Au3(P^P^P)2]3+ (P^P^P=Bis(dimethylphosphino-methyl)methylphosphin, dmmp).158, 161, 163, 182-184 Die für diese Beispiele mit durch Liganden eng zusammengehaltenen Metallatomen (Formel N) beobachteten, überraschenden photophysikalischen Eigenschaften gaben den Anstoß dafür, dass sich die mit der Excimer-Bildung verbundene Forschung zu einem unabhängigen, intellektuell lohnenswerten Forschungsobjekt entwickelt hat.
Frühe UV/Vis-Absorptionsstudien und Untersuchungen des Magnetozirkulardichroismus der zweikernigen AuI-dmpm- und -dmpe-Systeme in Lösung haben gezeigt, dass die zur Lumineszenz führenden Anregungen metallzentriert sind.161, 184 Bei der Zuordnung der Absorptions- und der prominentesten Lumineszenz-Banden verließen sich die meisten der frühen Autoren in Anlehnung an Gray und Mitarbeiter auf Symmetrieprinzipien und elektrostatische Ligandenfeld-ähnliche Betrachtungen zur Konstruktion von Molekülorbital-Diagrammen (MO) mit Metall-zentrierten HOMOs und LUMOs. Die HOMOs, gebildet durch Linearkombination hauptsächlich der 5d -Atomorbitale der beiden Goldatome (z-Achse entlang der Au-Au-Verbindungslinie in den schwach assoziierten Komplexen), wurden als dσ* bezeichnet. Die als pσ bezeichneten LUMOs entstanden durch Kombination von fast reinen 6pz-Atomorbitalen.
-Atomorbitale der beiden Goldatome (z-Achse entlang der Au-Au-Verbindungslinie in den schwach assoziierten Komplexen), wurden als dσ* bezeichnet. Die als pσ bezeichneten LUMOs entstanden durch Kombination von fast reinen 6pz-Atomorbitalen.
Fackler und Mitarbeiter158 führten an Modellkomplexen [Au2(H2PCH2PH2)2]2+ (N, R=H) mit Bis(phosphino)methan als Ligand in achtgliedrigen Ringen in verlängerter Sessel-Konformation SCF-Xα-SW Rechnungen mit relativistischen Korrekturen durch. Leider konnte die AuI⋅⋅⋅AuI-Assoziation mit diesen Untersuchungen nicht erklärt werden, und die relativen Energien der leeren Orbitale waren unzuverlässig. Die Ergebnisse stimmten jedoch mit mehr intuitiven Ansätzen überein. Trotz des Mangels solider theoretischer Belege stimmten die Autoren der Arbeiten158, 161, 163, 182-184 darin überein, dass die Absorptionen im UV-Bereich all dieser Komplexe Metall-zentrierten, Spin- und Laporte-erlaubten dσ*→pσ-Übergängen zuzuschreiben sind, mit einer Einelektronen-Anregung unter Einbeziehung zweier Singulett-Zustände (siehe Schema 7). Die von allen Komplexen gezeigte Emission (Phosphoreszenz) im sichtbaren Bereich hielt man im Wesentlichen für einen pσ→-d-Triplett-Singulett-Übergang. Che und Yam163, 183 plädierten auf qualitativer Basis kurzzeitig für eine Beteiligung von dγ*-Zuständen, z. B. d -basierten MOs an dem Übergang, akzeptierten aber, nachdem mehr experimentelle und theoretische Belege verfügbar waren, die allgemeine Ansicht eines dσ*→pσ-Anregungs- und eines pσ→dσ*-Emissionsprozesses.
-basierten MOs an dem Übergang, akzeptierten aber, nachdem mehr experimentelle und theoretische Belege verfügbar waren, die allgemeine Ansicht eines dσ*→pσ-Anregungs- und eines pσ→dσ*-Emissionsprozesses.

Mikrozustände und Termzustände für die MO-Konfigurationen dσ*2 und dσ*1pσ1 in Au2(P^P)22+.
Die Einelektronen-Anregung von einem antibindenden in ein bindendes MO führt formal zu einer Zunahme der Bindungsordnung auf eins und zur Ausbildung einer σ-Bindung zwischen den beiden Goldatomen. Die resultierende Molekülverzerrung (hauptsächlich die Verkürzung des Au-Au-Abstands) wird gemeinhin für die große (ca. 17 000 cm−1) Stokes-Verschiebung zwischen Absorptions- und Emissions-Maximum verantwortlich gemacht.158
Obwohl für den dreikernigen Komplex [Au3(dmpm)2]2+ (in Acetonitril) wieder eine Emission vom niedrig liegenden 3[dγ*1pσ1]-Niveau verantwortlich gemacht wird, impliziert diese angeregte Konfiguration immer noch eine AuI−AuI-Bindungssituation mit einer pz-pz-Orbital-Überlappung.185 Eine steigende Anzahl der PAuP-Einheiten führt zu einer engeren dσ*-pσ-Energielücke, wohingegen E(dσ*) zunimmt; [Aun+1]-Einheiten absorbieren bei niedrigerer Energie als [Aun]-Gruppen.163
 (1)
(1) (2)
(2)Das Standard-Elektrodenpotential des aktivierten Paars [Au2(dppm)23+]*/[Au2(dppm)22+]* wurde auf −1.1 V gegenüber einer gesättigten Kochsalz-Kalomel-Elektrode geschätzt. Yam und Mitarbeiter165 haben für das Redox-Paar [Au2(dmpm)23+]*/[Au2(dmpm)22+]* einen Wert von −1.7 V bestimmt.
 (3)
(3)In einem späteren Artikel derselben Gruppe wurde hauptsächlich eine kompetitive Exciplex-Bildung des aktivierten Phosphin-Komplexes mit dem Lösungsmittel für dessen Inaktivität in der C-H-Bindungsaktivierung verantwortlich gemacht.187
Parallel zu den oben beschriebenen Arbeiten wurden andere zwei- oder mehrkernige Au⋅⋅⋅Au-gebundene, lumineszierende Komplexe entdeckt und untersucht.188-191 In diesen Systemen waren Liganden an den Absorptions- und Emissions-Elektronentransfer-Prozessen beteiligt; der Bildung von kovalenten Au−Au-Bindungen wurde dabei keine besondere Beachtung geschenkt. Es ist jedoch zu erwarten, dass die Anregung von Elektronen von antibindenden HOMOs in d10-d10-Systemen in verfügbare, bindende LUMOs in unterschiedlichem Maße ebenfalls zu einer Stärkung der Au-Au-Bindung führen kann. In diesem Aufsatz konzentrieren wir uns im Wesentlichen auf Komplexe mit eindeutigen MC-Anregungen und Emissionen. Ligand- oder Metall- und Ligand-lokalisierte Übergänge werden nur dann im Detail berücksichtigt, wenn sie zu einer deutlichen Stärkung der Metall-Metall-Bindung führen (Abschnitt 4.2.4). Für andere Entwicklungen sei auf Übersichtsartikel von Gade,192 Bowmaker,193 Fackler,56 Yam194 und deren Mitarbeiter verwiesen.
Trotz vieler Forschungsaktivitäten waren gegen Ende des 20. Jahrhunderts keine nennenswerten experimentellen oder theoretischen Hinweise auf oder Informationen über die AuI−AuI-Bindung im angeregten Zustand von doppelt verbrückten Diphosphin-Komplexen des Golds verfügbar. Bald darauf berichteten Che und Mitarbeiter, darunter hervorzuheben sind Phillips und Zhang, über wichtige Fortschritte in den Kenntnissen von den angeregten Zuständen sowie der molekularen Dynamik aktivierter Komplexe.
4.2.2 Weiterführende experimentelle und theoretische Untersuchungen
 (4)
(4) (5)
(5)Wie bereits erwähnt, macht die Exciplex-Bildung zwischen Excimeren 3[Au2(P^P)22+]* und dem Solvens solche Komplexe weniger gut zugänglich für Aktivierung von C-H-Bindungen, wenn die Wechselwirkung einem “Inner-sphere”-Mechanismus folgt.187 Eine Untersuchung der Reaktivität von Au−Au-gebundenen [Au2(dpim)2]2+-Komplexen (dpim=2-(Diphenylphosphino)-1-methylimidazol) im angeregten Zustand hat in Übereinstimmung mit den Exciplex-Vorschlägen von Che196 die Abhängigkeit der Emissionsenergie von der Fähigkeit des Gegenions zur Koordination bestätigt.
Die linearen dreikernigen Komplexe [Au3(dcpm)2]Y3 (Y=ClO4−, PF6−, CF3SO3−, Cl, SCN−) zeigen im Festkörper und in CH2Cl2-Lösung ziemlich genau dasselbe Relaxationsverhalten wie ihre oben beschriebenen zweikernigen Homologe.197 Sowohl die höherenergetischen intrinsischen Banden als auch die Emissionen niedrigerer Energie – letztere werden der Exciplex-Bildung mit den koordinierenden Gegenionen Cl− and SCN− zugewiesen – sind für dreikernige Komplexe jedoch rotverschoben und werden in den Wellenlängenbereichen 442–452 bzw. 558–634 nm gefunden. DFT-Berechnungen stützen ein früheres Ergebnis,163 demzufolge Emissionsenergien in mehrkernigen Komplexen mit linearen Ketten eine inverse Beziehung zur Anzahl der Metallatome in der Kette aufweisen. Emissionsenergien unterhalb von 500 nm in linearen mehrkernigen Goldkomplexen sollten nicht auf der Basis einer Au−Au-Exciplex-Bindung erklärt werden. Bemerkenswerterweise wird bei allen Prozessen in Lösung kein C-X-Bindungsbruch in Alkylhalogeniden als Lösungsmittel beobachtet (siehe unten).
Che und Mitarbeiter152 haben über eine Raman-spektroskopische Untersuchung der Verbindung [Au2(dcpm)2](ClO4)2 in Lösung berichtet, die auf die Zuordnung der Dipol-erlaubten Absorptionsbande mit der niedrigsten Energie bei ca. 277 nm ausgerichtet war, unter Einbeziehung des allgemein akzeptierten angeregten Zustands 1[dσ*pσ]. Über verschiedene Formalismen und die Condon-Näherung konnte gezeigt werden, dass die vermutete Anregung dσ*→pσ mit den Raman-spektroskopischen Ergebnissen konsistent ist. Die Veränderung der Au-Au-Bindungslänge beim Übergang vom Singulett-Grundzustand (oder dem ursprünglichen Franck-Condon-Produkt) in den ersten Schwingungs-relaxierten, angeregten Singulett-Zustand beträgt 0.11 Å, was zu einer Bindungslänge von 2.81 Å für das doppelt verbrückte Excimer führt. Die im Wesentlichen durch p-p-Überlappung gebildete AuI−AuI-σ-Bindung ist etwas länger als der Wert von ca. 2.6 Å, um welchen die Goldatome in den zweikernigen Au0- und AuII-Komplexen auseinander liegen, für die ein größerer Beitrag der s-Orbitale an der Bindung gefordert wird.7 Alle weiteren Diskussionen in diesem Abschnitt beziehen sich lediglich auf elektronische Grund- und angeregte Zustände; Schwingungs- und Rotations-Energieniveaus werden nicht berücksichtigt.
Zum Vergleich durchgeführte Absorptions- und Resonanz-Raman-spektroskopische Untersuchungen an [M2(dcpm)2]2+ Komplexen von Kupfer, Silber und Gold haben bestätigt, dass die M-M-Bindungen aller drei Komplexe während der MC-Anregung stärker werden.198-200 Die mit dem Singulett-Singulett-Übergang verbundene Veränderung der Bindung folgt der Reihenfolge ΔCu−Cu>ΔAg−Ag>ΔAu−Au. Im Fall der Gold-Komplexe wird die Bindungsverstärkung offensichtlich durch Lösungsmittel-Komplexierung im aktivierten Zustand begünstigt, wohingegen die Koordination durch einen dritten starken Donor-Liganden wie in Komplexen der Zusammensetzung [M2(dmpm)3]2+, in denen die Koordinationszahl von CN=2 auf CN=3 gestiegen ist, sowohl im Grund- als auch im angeregten Zustand eine Verlängerung oder sogar den Bruch der Au-Au-Bindung bewirkt.
In den frühen 2000er Jahren sind von Che und Zhang153, 201 sowie Zhang und Pan166, 202 zwei wichtige theoretische Publikationen über Komplexe des Typs [Au2(P^P)2]2+ und [Au2(P^S)2]2+ erschienen. In den Rechnungen dienten Diphosphinomethan (H2PCH2PH2, dpm), C-Mercapto-phosphinomethan (HSCH2PH2, mpm) und C-(Methylsulfido)phosphinomethan (MeSCH2PH2, mspm) als Modelle für neutrale, ditopische Diphosphin- und Phosphinothioether-Liganden in den entsprechenden zweikernigen, metallacyclischen, alle in der Sesselform vorliegenden Goldkomplexen.201 Man beachte, dass die freien Elektronenpaare der Liganden in die 6s- und 6p-Orbitale der unkoordinierten [Au2]2+-Systeme eingebracht werden und damit die formal geschlossenschalige 5d106s0-Konfiguration jedes involvierten Goldatoms modifiziert wird, damit weitere Bindungskräfte wirksam werden können. Diese Betrachtung in Kombination mit London'schen Dispersionskräften erfordert zur korrekten Beschreibung der aurophilen Wechselwirkung im Grundzustand wie bereits früher erwähnt den Einsatz von Methoden, die die Konfigurationswechselwirkungen mit einschließen. Zur Optimierung der Strukturen im Grundzustand verwendeten die Autoren eine MP2-basierte Näherung. Für die Beschreibung der angeregten Zustände wurde die CIS-Methode (configuration interaction singles) eingesetzt, die vermutlich ausreicht, angesichts der wichtigeren Rolle der Orbital-Überlappung im Vergleich zur Situation im Grundzustand. Zur theoretischen Nachahmung der Rolle von Lösungsmittelmolekülen bzw. Gegenionen wurden getrennte Rechnungen für die Modell-Komplexe [Au2(dpm)2(CH3CN)2]2+, [Au2(mpm)2(CH3CN)2]2+, [Au2(mspm)2(CH3CN)2]2+, [Au2(dpm)2](ClO)2 und [Au2(mpm)2](ClO)2 durchgeführt, wobei die letzten beiden das Verhalten der Komplex-Typen [Au2(P^P)2](ClO4)2 und [Au2(P^S)2](ClO4)2 im festen Zustand simulieren sollten. Die von Che und Mitarbeitern gemachten Vorschläge bezüglich der Anwesenheit und Position von zwei emittierenden Triplett-Zuständen stellten sich als realistisch heraus. Die relativen Term-Energien für die vorgeschlagenen angeregten Zustände des Excimers [Au2(dcpm)22+]* und seiner durch Zusammenlagerung mit Lösungsmittel-Molekülen entstandenen Exciplexe wurden in diesen Modelluntersuchungen gut reproduziert.
Die Ergebnisse theoretischer Untersuchungen ausgewählter [Au2(P^P)2]2+-Komplexe haben gezeigt, dass die Anregung im UV-Bereich Metall-zentriert ist, was zu einer Elektronenkonfiguration 5dσ*16pσ1 führt. Die Ausbildung einer kovalenten Bindung im angeregten Zustand wird zunächst durch Überlappung der pz-Atomorbitale erreicht. Es werden zwei langlebige Triplett-Zustände niedrigerer Energie vorhergesagt, wobei der energiereichere von beiden (der intrinsische Zustand eines nicht-solvatisierten Komplexes) im nahen UV-Bereich emittiert und einer Elektronenkonfiguration 5dσ*1(sp)σ1 entspricht. Die daraus resultierende stärkere Au−Au-Bindung wird durch Überlappung erheblich hybridisierter 6s- und 6p-Atomorbitale gebildet. Die linearkombinierten 6s-Orbitale sind nur an der Bildung der Au−Au-Bindung für den Triplett-Zustand niedrigster Energie, 3[5dσ*16sσ1], beteiligt, was das Auftreten kurzer Au−Au-Bindungen erklärt. Dieser Zustand entspricht der Bildung von Exciplexen aus Komplex-Lösungsmittel- und/oder Komplex-Gegenion-Wechselwirkung. Insgesamt ist die auf der Basis theoretischer Berechnungen vorhergesagte, während der elektronischen Anregung stattfindende Bindungsverkürzung viel deutlicher als über Raman-Experimente abgeschätzt wurde. Nach Che und Mitarbeitern195 findet die nicht-solvatisierte Situation ihre praktische Entsprechung in kristallinen, doppelt verbrückten Bis(phosphin)-Komplexen, in denen sich das Gegenion weit entfernt von den Metallzentren aufhält. Für Fälle, in denen elektrostatische Kation-Anion-Wechselwirkungen wichtig werden, zeigen theoretische Modelle eine Stärkung der Au−Au-Wechselwirkung und eine Rotverschiebung der Emission im festen Zustand, ähnlich der Situation, wie sie für das Aggregat nach Zugabe von Lösungsmittel zum Modellkomplex berechnet wurde. Gleichzeitig zur Stärkung der Au−Au-Wechselwirkung durch Excimer- und Exciplex-Bildung werden die Metall-Ligand-Bindungen (Au−P und Au−S der Phosphin-, Thiol- bzw. Thioether-Donoren) geschwächt und verlängert. In Schema 8 sind die Absorptions- und Emissions-Ergebnisse für [Au2(dcpm)2]2+ mit den für verwandte Modellkomplexe erhaltenen theoretischen Informationen in einem Jablonski-Diagramm kombiniert, in dem die Änderung der [Au−Au]*-Bindungslänge gegen die Reaktionskoordinate aufgetragen ist. Wir weisen darauf hin, dass die Emission mit der niedrigsten Energie in Modellkomplexen des allgemeinen Typs [Au2(HSCH2PH2)2]2+ eine kleine MMLCT-Komponente enthält.

Qualitatives, stark idealisiertes Jablonski-Energie-Zustandsdiagramm mit den verschiedenen, für Absorption und Emission von [Au2(dcpm)2]Y2 in Lösung und im festen Zustand verantwortlichen Spezies (A=Absorption; P=Phosphoreszenz).
Nach einer kürzlich durchgeführten, ultraschnellen, zeitaufgelösten transienten Absorptions- (TRTA) und zeitaufgelösten Emissions (TRE)-spektroskopischen Untersuchung203 bildet sich das intrinsische Excimer im Triplett-Zustand sofort (d. h. innerhalb von 0.15 ps) bei Bestrahlung von [Au2(dcpm)2](ClO4)2 in einem ultraschnellen Prozess ohne erkennbares Eingreifen vom Lösungsmittel. Der Triplett-Zustand zerfällt mit einer Lebensdauer von 4.3 μs in den Grundzustand. Wird der Goldkomplex in CH3CN gelöst, kommt es durch Komplexbildung mit Lösungsmittel-Molekülen schnell zur Exciplex-Bildung (Zeitkonstante 4.5 ps). Dieser Vorgang zeigt die Neigung aktivierter AuI-Komplexe zur Aufweitung ihrer Koordinationszahl auf CN>2. Mit CH2Cl2 als Lösungsmittel ist das Ergebnis ein völlig anderes (nicht gezeigt in Schema 2), und der inhärente Triplett-Zustand wird durch C-X-Bindungsbruch [vergleiche Gl. (3)] binnen 510 ps zu einem phosphoreszierenden Triplett-Intermediat unbekannter Zusammensetzung umgewandelt. Zeitskalen und Strukturinformationen zur Exciplex-Bildung durch Bindung von Gegenionen [Gl. (4)] stehen noch aus.
Während in AuI⋅⋅⋅AuI-assoziierten Komplexen die Verkürzung der Au-Au-Bindung die wichtigste Verzerrung im angeregten Zustand ist, reorganisieren sich Komplexe des Typs Au(PR3)3 während der Anregung durch Symmetrieänderung von trigonal nach T-förmig.204 Dieses theoretische Ergebnis widerspricht früheren Interpretationen, nach denen große Stokes-Verschiebungen in solchen dreifach koordinierten Komplexen auf eine Kontraktion der Au-Liganden-Bindung zurückgehen.205, 206
4.2.3 Ähnliche Beobachtungen in zwei- und dreikernigen AuI-Komplexen mit verbrückenden Phosphin-Carben-Liganden
Ein unsymmetrischer, dimetallacyclischer Komplex mit Carben/Phosphin-funktionalisierten Liganden wurde kürzlich von Danopoulos, Braunstein und Mitarbeitern beschrieben.168 Die einzige wichtige Besonderheit ihres P^C^P-Liganden bzgl. seines Koodinations- und spektrochemischen Verhaltens sind die beiden exo-ständigen Phosphingruppen, eine davon frei hängend in P, während an der linear dreiatomigen Au3-Kette alle drei Donor-Funktionen koordinativ beteiligt sind (Q
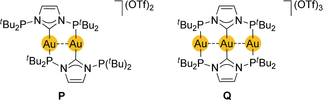 ). Diese Struktur wird beim Beispiel [Au3L2](OTf)3 gefunden, welches an die von Yam und Wong untersuchten194 Komplexe vom Typ [Au3(dmpm)2]3+ und an ein von Deák und Mitarbeitern beschriebenes gemischtes Komplexsalz erinnert.207 Nur Verbindung Q zeigt eine intensive, fast reine MC-Absorptionsbande in Lösung (λmax=345 nm) neben anderen Banden höherer Energie, an denen die Ligand-Orbitale beteiligt sind. Die wichtige Rolle der dσ*1pσ1-Konfiguration und einer signifikanten kovalenten Au−Au-Bindung im angeregten Zustand wurde anhand von Resonanz-verstärkten Raman-Spektren demonstriert. Eine Verfolgung der MC-Übergänge zeigt die Schwingungen der kovalenten Au−Au-Bindung im angeregten Zustand. Hier konzentrieren wir uns hauptsächlich auf die Ergebnisse für Q. Diese Verbindung zeigt eine schmale, intensive blau-violette Strahlungsbande bei 446 nm, die wesentlich von der Polarität des Lösungsmittels, dem Zustand (Lösung, Glasmatrix, reines Pulver) und der Temperatur sowie von der Natur des MC 3[5dσ*6pσ]-Triplett-Zustands abhängig ist.197 Die Ergebnisse theoretischer Untersuchungen (auf DFT-Niveau) haben gezeigt, dass für P im angeregten Zustand die Koordination der frei hängenden Phosphin-Gruppe zur Stabilisierung des emittierenden Triplett-Zustands beitragen könnte. Die Abnahme der Au-Au-Abstände im angeregten Zustand in Q ist trotz der starren Liganden eindeutig. Ein quantitativer Vergleich der Abstände in Grund- und angeregtem Zustand ist in Anbetracht der Unzulänglichkeiten von DFT-Berechnungen für den Grundzustand nicht wirklich sinnvoll.7, 208 Bemerkenswerterweise verändern sich die Au-P- und Au-C-Abstände während der Anregung nur sehr wenig.
). Diese Struktur wird beim Beispiel [Au3L2](OTf)3 gefunden, welches an die von Yam und Wong untersuchten194 Komplexe vom Typ [Au3(dmpm)2]3+ und an ein von Deák und Mitarbeitern beschriebenes gemischtes Komplexsalz erinnert.207 Nur Verbindung Q zeigt eine intensive, fast reine MC-Absorptionsbande in Lösung (λmax=345 nm) neben anderen Banden höherer Energie, an denen die Ligand-Orbitale beteiligt sind. Die wichtige Rolle der dσ*1pσ1-Konfiguration und einer signifikanten kovalenten Au−Au-Bindung im angeregten Zustand wurde anhand von Resonanz-verstärkten Raman-Spektren demonstriert. Eine Verfolgung der MC-Übergänge zeigt die Schwingungen der kovalenten Au−Au-Bindung im angeregten Zustand. Hier konzentrieren wir uns hauptsächlich auf die Ergebnisse für Q. Diese Verbindung zeigt eine schmale, intensive blau-violette Strahlungsbande bei 446 nm, die wesentlich von der Polarität des Lösungsmittels, dem Zustand (Lösung, Glasmatrix, reines Pulver) und der Temperatur sowie von der Natur des MC 3[5dσ*6pσ]-Triplett-Zustands abhängig ist.197 Die Ergebnisse theoretischer Untersuchungen (auf DFT-Niveau) haben gezeigt, dass für P im angeregten Zustand die Koordination der frei hängenden Phosphin-Gruppe zur Stabilisierung des emittierenden Triplett-Zustands beitragen könnte. Die Abnahme der Au-Au-Abstände im angeregten Zustand in Q ist trotz der starren Liganden eindeutig. Ein quantitativer Vergleich der Abstände in Grund- und angeregtem Zustand ist in Anbetracht der Unzulänglichkeiten von DFT-Berechnungen für den Grundzustand nicht wirklich sinnvoll.7, 208 Bemerkenswerterweise verändern sich die Au-P- und Au-C-Abstände während der Anregung nur sehr wenig.
4.2.4 Der Einfluss von Heteroatomen auf die AuI-AuI-Bindung
Lumineszierende, zweikernige Phosphin-verbrückte Gold(I)-Komplexe, die auch noch Thiolat-Liganden (einfach verbrückt, Form R; doppelt verbrückt, Form S
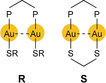 ) enthalten, und auch einfachere, einkernige Komplexe vom Typ R3PAuSR′ wurden von verschiedenen Forschungsgruppen experimentell untersucht.209-213 In allen diesen Beispielen sind die freien (als S(pπ) bezeichneten) Elektronenpaare des S-Donors eng an den Anregungs- und Emissions-Prozessen und schließlich an der [Au−Au]*-Bindungsbildung beteiligt. Unter für eine Au⋅⋅⋅Au-Bindung günstigen, idealisierten Voraussetzungen befinden sich die beiden Gold-Atome in enger Nachbarschaft zueinander, und die Erstanregung niedrigster Energie wird qualitativ als S(pπ)→pσ-Elektronenübergang (oder Ligand-zu-Metall-Metall-Charge-Transfer, LMMCT)212, 213 beschrieben, wobei es sich bei pσ um das bindende LUMO, entstanden durch die Überlappung lokalisierter pz-Orbitale in der Au2-Einheit, handelt.209, 211, 212 Strukturinformationen über die Relaxationsdynamik der resultierenden 1[LMMCT]*- und 3[LMMCT]*-Zustände und damit über die progressive Excimer- oder Exciplex-Bildung liegen noch nicht vor.
) enthalten, und auch einfachere, einkernige Komplexe vom Typ R3PAuSR′ wurden von verschiedenen Forschungsgruppen experimentell untersucht.209-213 In allen diesen Beispielen sind die freien (als S(pπ) bezeichneten) Elektronenpaare des S-Donors eng an den Anregungs- und Emissions-Prozessen und schließlich an der [Au−Au]*-Bindungsbildung beteiligt. Unter für eine Au⋅⋅⋅Au-Bindung günstigen, idealisierten Voraussetzungen befinden sich die beiden Gold-Atome in enger Nachbarschaft zueinander, und die Erstanregung niedrigster Energie wird qualitativ als S(pπ)→pσ-Elektronenübergang (oder Ligand-zu-Metall-Metall-Charge-Transfer, LMMCT)212, 213 beschrieben, wobei es sich bei pσ um das bindende LUMO, entstanden durch die Überlappung lokalisierter pz-Orbitale in der Au2-Einheit, handelt.209, 211, 212 Strukturinformationen über die Relaxationsdynamik der resultierenden 1[LMMCT]*- und 3[LMMCT]*-Zustände und damit über die progressive Excimer- oder Exciplex-Bildung liegen noch nicht vor.
Strukturuntersuchungen in Kombination mit Lumineszenz-spektroskopischen Studien haben als wichtiges Ergebnis gezeigt, dass die Stärke der aurophilen Au⋅⋅⋅Au-Wechselwirkung im Grundzustand in verschiedenen Phosphin-Thiolat-Komplexen von AuI nicht notwendigerweise der dominierende Faktor in der Emissionsenergetik im festen Zustand ist, noch dass sie immer direkt mit diesen Energien korreliert. Die Zusammensetzung und räumliche Orientierung der Thiol-Liganden kann beispielsweise genauso wichtig sein oder sogar eine entscheidende Rolle spielen.210, 211 Zhang und Pan214, 202 haben die Orbitalbeteiligung bei optischen Übergängen in drei neutralen Modellkomplexen, [Au2(dpm)(SCH2S)], [Au2(H2PCH2S)2] und [Au2(HSCH2S)2], mit MP2- und CIS-Methoden berechnet. Ihre Ergebnisse bestätigen die oben gegebene Interpretation bezüglich der Au-Au-Bindungsbildung und -stärkung im angeregten Zustand nach dem Elektronenübergang S(pπ)→sσ und/oder pσ. Obwohl die Zuordnung der Emission mit niedrigster Energie in den drei Komplexen nicht so einfach ist, so stammt sie vermutlich doch von Metall-Metall-lokalisierten Triplett-Zuständen.
Auf welche Art und Weise weiche Heteroatom-Donor-Liganden zur Bildung einer kovalenten Au−Au-Bindung während einer UV/Vis-Anregung beitragen, wurde durch die Ergebnisse von Saillard, Liu, Boucekkine und Mitarbeitern geklärt.215, 216 Die Autoren untersuchten zweikernige Diseleno- und Dithiophosphinat-Komplexe T und U
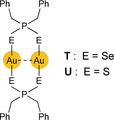 mittels experimenteller (Einkristall-Röntgenbeugung, Anregungs-, Emissions- und Raman-Spektroskopie) und rechnerischer (zeitabhängige DFT) Methoden.
mittels experimenteller (Einkristall-Röntgenbeugung, Anregungs-, Emissions- und Raman-Spektroskopie) und rechnerischer (zeitabhängige DFT) Methoden.
Sehr schwache Au⋅⋅⋅Au-Wechselwirkungen mit intramolekularen Abständen von 3.387 und 3.456 Å für T und 3.267 Å für U sind charakteristisch für diese Komplexe. Die strukturlosen Anregungs- und Emissionsbanden niedrigster Energie haben bei tiefen Temperaturen (77 K) in festem 2-MeTHF ihre Maxima λmaxexc und λmaxem bei 434 bzw. 593 nm für T und bei 471 bzw. 565 nm für U. Die Banden der Lösungsspektren sind alle hypsochrom verschoben. In erster Näherung können die Anregungs- und Emissions-Prozesse wieder einem Elektronenübergang zwischen dem HOMO und LUMO zugeschrieben werden. Erstere Orbitale der Energie E(pπ) entstehen durch Linearkombination von mit freien Elektronenpaaren von Se und S gefüllten Orbitalen, die aber auch 12–16 % Au-d-Orbital-Charakter besitzen. Das LUMO von T wird von Au-p-Orbital-Anteilen dominiert, während das LUMO von U zu 35 % Goldanteil (d-Orbitale) besitzt, ergänzt durch 46 % Schwefelcharakter.
Während der Anregung wird Elektronendichte von einem nicht-bindenden in ein zwischen zwei Goldatomen lokalisiertes, bindendes MO übertragen; die als σ(p–p) beschriebene Überlappung ist für die Bildung der Au−Au-Bindung verantwortlich (Bindungsordnung ≤0.5). Die Ergebnisse von Resonanz-Raman-spektroskopischen Untersuchungen bestätigen eindeutig Au-Au-Streckbewegungen in den resultierenden angeregten Singulett- und Triplett-Zuständen, die in den Grundzuständen völlig fehlen. DFT-Rechnungen zufolge verringert sich der Abstand zwischen den Goldatomen im angeregten Triplett-Zustand gegenüber dem Grundzustand um bis zu 25 %. Diese Ergebnisse liefern überzeugende Belege dafür, dass nicht nur die aurophile Au⋅⋅⋅Au-Wechselwirkung die AuI-AuI-Bindungsbildung positiv beeinflusst, sondern dass weiche Heteroatomdonor-Liganden ebenfalls eine wichtige Elektronenquelle für eine derartige Bindung darstellen können. Andererseits unterschätzt die vorgeschlagene LMMCT-Beschreibung für den Anregungsprozess die beträchtliche Beteiligung der Heteroatom-Orbitale an den LUMO-Akzeptor-Orbitalen. NBO-Analysen sagen eine Rehybridisierung entlang der Reaktionskoordinate vorher.
4.3 Aus einkernigen AuI-Komplexen entstandene zwei- und mehrkernige Excimere
4.3.1 Der Fall Dicyanidoaurat(I)
Verbindungen mit dem Anion [Au(CN)2]− sind erstklassige Beispiele und Prototypen für lineare, einkernige AuI-Komplexe, die bei Anregung intermolekulare Excimere und Exciplexe bilden. Verschiedene frühe spektrophotometrische Untersuchungen haben ergeben, dass Lösungen dieser Verbindungen mit Konzentrationen von ≥0.01 m Photolumineszenz zeigen. Dieses Ergebnis wurde einer Oligomerisierung in Lösung zugeschrieben, einem Prozess, der auf kooperative aurophile Wechselwirkungen zurückzuführen ist (siehe oben).101
In einer wichtigen, richtungsweisenden Veröffentlichung haben Patterson, Fackler und Mitarbeiter107 die Ergebnisse einer Untersuchung dieses Lumineszenz-Phänomens beschrieben, bei der sie sich auf Wechselwirkungen in angeregten Zuständen in Lösung konzentrierten. Wenngleich in der Regel in wässriger Lösung bei Raumtemperatur nur zwei größere unstrukturierte Emissionsbanden mit großer Stokes-Verschiebung beobachtet werden, so können doch durch Veränderungen von Konzentration, Lösungsmittel, Temperatur und Anregungs-Wellenlänge eine Vielzahl von Banden erzeugt und getrennt werden. Generell bilden schwach assoziierte Au-Au-Dimere oder Oligomere bei selektiver Anregung die entsprechenden Excimere; verwandte Exciplexe höherer Ordnung entstehen, wenn solche Excimere mit zusätzlichen, im Grundzustand befindlichen, monomeren Komplexeinheiten wechselwirken und sich an diese binden. Beginnend mit der Emission höchster Energie, welche einem Triplett-Zustand des Excimers [Au(CN)2−]n* mit kleinster Nuklearität (n=2) zugeordnet wird, gehören die Banden mit zunehmend geringerer Energie zu wachsenden Oligomeren (n>2) (Exciplexen), die durch sukzessive Addition von Monomeren an die oligomere Kette entstanden sind (Formeln I, J, siehe oben).
Im Fokus der Untersuchungen standen die Veränderungen der Au-Au-Bindungslänge und -stärke, die parallel zu den spektroskopischen Übergängen von höher energetischen Emissionsbanden zu solchen niedrigerer Energie ablaufen. Es ist zu beachten, dass die Exciplex-Bildung unter Beteiligung des Lösungsmittels, wie früher für doppelt verbrückte (Phosphin)digold-Komplexe im angeregten Zustand angenommen, ausgeschlossen werden kann, weil sich die Emissionsbanden in Lösung und im festen Zustand in ihrer (ursprünglichen) Form und Energie gleichen. Einige der oben erwähnten Annahmen und Vorschläge wurden in derselben Publikation mit theoretischen Methoden auf MP2-Basis an [AuCN)2−]2 in der gestaffelten Dimer-Konformation als Modell genauer untersucht (Formel I, siehe oben). Während der Anregung und Excimer-Bildung nimmt der berechnete Au-Au-Abstand von 2.96 auf 2.66 Å ab. Gleichzeitig kommt es fast zur Verdopplung der berechneten aktiven Streckfrequenz νAu−Au, und die Kraftkonstante wächst um eine Größenordnung. Die Länge der kovalenten Au−Au-Bindung im angeregten Zustand ist wieder nicht viel größer als in den wohlbekannten, nicht Ligand-gestützten und nur über Gold-Gold-Einfachbindungen verknüpften AuII-AuII-Komplexen (ca. 2.6 Å, siehe oben). Extended-Hückel-Berechnungen des angeregten Zustands zeigen eindeutig, dass die [AuI−AuI]*-Bindungen mit zunehmender Oligomerisierung innerhalb der Reihe [Au(CN)2−]n stärker werden: Das Exciplex [Au(CN)2−]3* beispielsweise ist stärker gebunden (niedrigere Triplett-Energie) als das Excimer [Au(CN)2−]2*. Außerdem wird die Au−Au-Bindung von der Geometrie (linear, gewinkelt) und der Konformation (ekliptisch, gestaffelt) des jeweiligen Exciplexes beeinflusst. Auf diese Aspekte wird weiter unten näher eingegangen. Obwohl der [Ag(CN)2]−-Komplex photochemisch im Allgemeinen seinem höheren Homologen ähnlich ist, so sind interessanterweise die aurophilen Au⋅⋅⋅Au-Wechselwirkungen im Grundzustand vergleichsweise stärker als die argentophilen Ag⋅⋅⋅Ag-Wechselwirkungen, während in den entsprechenden angeregten Zuständen die Ag−Ag-Wechselwirkungen stärker sind als die Au−Au-Wechselwirkungen.
Die erste Lumineszenz-Untersuchung an reinen Einkristallen von K[Au(CN)2] wurde 1986 im Labor von Patterson durchgeführt.95 Obwohl die Untersuchungen und Diskussionen auf Variationen der Au-Au-Atomüberlappung im Grundzustand fokussiert waren, bildeten die Ergebnisse das Fundament für den später entwickelten sogenannten Tuning-Ansatz. Excimer- und Exciplex-Tuning bezieht sich auf die selektive Generierung von separierten Emissions-Banden für ein ausgewähltes lumineszierendes System über die Variation verschiedener, unten erklärter, experimenteller Parameter. Die Forschungsrichtung des Tuning erhielt in den letzten Jahren viel Aufmerksamkeit aufgrund möglicher optochemischer Anwendungen in der Energie-Übertragung durch Excitonen,217 was sich zur Photokatalyse – sogar in Großprozessen wie der Zersetzung von Pestiziden zur Dekontamination von Industrieabwässern – eignen könnte.218 Signifikantes Tuning wurde für [Au(CN)2−]n* in reinem K[Au(CN)2] oder in dotierten K[Au(CN)2]/KCl-Kristallen erreicht.105
Die Assoziation der Anionen in kristallinem K[Au(CN)2] und Cs[Au(CN)2] führt zur Bildung von Schichten aus Goldatomen (Abbildung 1).98 In erster Näherung wird die elektronische Situation innerhalb der Kristalle im Sinne von Elektronenkonfigurationen mittels Einelektronen-Energiebändern beschrieben. Das Valenzband (VB) besteht hauptsächlich aus den 5d - und 6s-Orbitalen der beteiligten Au-Atome und das Leitungsband (CB) im Wesentlichen aus 6pz-Orbitalen, gemischt mit kleinen Beiträgen von Ligand-Orbitalen. Spektroskopische Übergänge werden dann einfach als Elektronentransport von VB nach CB oder umgekehrt beschrieben. Eine vollständigere Beschreibung der spektroskopischen Zustände erfordert neben anderen Aspekten eine Berücksichtigung von Elektron-Elektron- (Term-Generierung), Loch-Elektron- (Bildung delokalisierter Excitonen) und Exciton-Gitter-Wechselwirkungen (Erzeugung von lokalisierten “self-trapped” Zuständen). Exciton-Gitter-Wechselwirkungen zweiten Grades führen zu einer Verkürzung des Au−Au-Abstands und einer stärkeren Bindung innerhalb der Ketten oder Schichten aus Goldatomen. Eine ausführliche theoretische Beschreibung für die [Au(CN)2]−-Schichten in M[Au(CN)2]-Kristallen liegt bisher nicht vor, wurde aber für quasi-eindimensionale Verbindungen Mn[Pt(CN)4]⋅x H2O (M=Na, K, Ba, Mg, …; n=2 oder 1) vorweggenommen.219
- und 6s-Orbitalen der beteiligten Au-Atome und das Leitungsband (CB) im Wesentlichen aus 6pz-Orbitalen, gemischt mit kleinen Beiträgen von Ligand-Orbitalen. Spektroskopische Übergänge werden dann einfach als Elektronentransport von VB nach CB oder umgekehrt beschrieben. Eine vollständigere Beschreibung der spektroskopischen Zustände erfordert neben anderen Aspekten eine Berücksichtigung von Elektron-Elektron- (Term-Generierung), Loch-Elektron- (Bildung delokalisierter Excitonen) und Exciton-Gitter-Wechselwirkungen (Erzeugung von lokalisierten “self-trapped” Zuständen). Exciton-Gitter-Wechselwirkungen zweiten Grades führen zu einer Verkürzung des Au−Au-Abstands und einer stärkeren Bindung innerhalb der Ketten oder Schichten aus Goldatomen. Eine ausführliche theoretische Beschreibung für die [Au(CN)2]−-Schichten in M[Au(CN)2]-Kristallen liegt bisher nicht vor, wurde aber für quasi-eindimensionale Verbindungen Mn[Pt(CN)4]⋅x H2O (M=Na, K, Ba, Mg, …; n=2 oder 1) vorweggenommen.219
Die Beeinflussung der breiten Emissionsbanden (bei 397 bzw. 435 nm) von K[Au(CN)2] und Cs[Au(CN)2] durch äußere Einwirkungen wurde ebenfalls gemessen.98 Unter hydrostatischem Druck (bis zu 40 kbar) werden in linearer Abhängigkeit große Rotverschiebungen von −300 cm−1 bzw. −200 cm−1 kbar−1 beobachtet. Die Verschiebungen hängen ganz offenbar mit der relativen Kompressibilität der Kristalle zusammen. Auf Mikroebene führt die Druckerhöhung zur Verringerung der Au−Au-Bindungslänge im angeregten Zustand im Vergleich zum Grundzustand; die Bandlücke schließt sich und die Bandenergie ist erniedrigt. Die breite Emissionsbande von Cs[Au(CN)2] kann bei 5 K in zwei Hauptbanden aufgelöst werden. Die Bande niedrigerer Energie (454 nm) hat die Eigenschaften eines lokalisierten Zustands, an dem sehr wahrscheinlich ein Cluster aus nur wenigen [Au(CN)2]−-Komplexeinheiten beteiligt ist, wie dies schon für die Mn[Pt(CN)4]-Beispiele gefunden wurde. Die Bande höherer Energie entspricht den delokalisierten Clusterzuständen {[Au(CN)2]n}n−. Der Zusammenhang der Emissionsenergien mit den Au-Au-Abständen in Kristallen ist vergleichbar mit Effekten, die für die Exciton-Gitter-Wechselwirkung, beispielsweise in eindimensionalen Anordnungen komplexer Anionen in Kristallen von Ba[Pt(CN)4]⋅4 H2O, beobachtet wurden.220
Eine erhöhte Dotierung mit K[Au(CN)2] in KCl-Kristallen führt zu höheren Oligomeren mit einer stärkeren Au−Au-Bindung im angeregten Zustand und als Folge zu rotverschobenen Emissionsbanden.103 Verschiedene Struktur-Versionen eines gegebenen Oligomers beeinflussen auch die Stärke der Au⋅⋅⋅Au-Wechselwirkungen, wie in anderem Zusammenhang von Schmidbaur gezeigt wurde.221 Der Effekt der Kontraktion ist im angeregten Zustand stärker ausgeprägt als im Grundzustand. Außerdem kann durch Variation der Anregungswellenlänge für den jeweiligen Kristall eine steigende Zahl an Emissionsquellen erzeugt werden, die langlebigen angeregten Zuständen von mindestens drei Exciplexen entsprechen.
Die Anzahl und Frequenzen der Raman-Banden für kristalline, [Au(CN)2]−-dotierte Materialien korrelieren deutlich mit den Photolumineszenz-Banden der unterschiedlich hoch dotierten Kristalle aus einem Satz von Proben. Dieses Ergebnis zeigt an, dass die [Au(CN)2−]n-Ionen im Kristall abhängig vom Wert n in unterschiedlichen Umgebungen angetroffen werden. Jedes der Oligomere im Grundzustand und jedes der entsprechenden während der Anregung gebildeten Exciplexe geht eine Au−Au-σ-Einfachbindung ein, was sich jeweils in einer Emissionsbande vom angeregten Triplett-Zustand niederschlägt. Es sollte im Auge behalten werden, dass zwei oder mehrere Emissionsbanden überlappen können, wenn mehr als ein Exciplex mit ziemlich ähnlicher Bindungsenergie vorhanden ist.
Ein breites Exciplex-Tuning (in einem Bereich von 22000 cm−1) mit selektiven Veränderungen der Au-Au-Bindungen kann über eine Variation des Mediums (reine Kristalle, dotierte Kristalle, Lösungen), der Temperatur, der Komplexkonzentration, des Gegenions und des verwendeten Wirtsgitters erreicht werden.106 Die jeweiligen Abbildungen 9 in den Referenzen 103 und 107 stellen die verblüffenden, bisher diesbezüglich erzielten Erfolge heraus. Im Rahmen dieser Übersicht bezieht sich der Begriff “Tuning” nicht nur auf die erfolgreiche Auftrennung von Emissionsbanden nach abnehmender Energie, sondern es liefert gleichzeitig ein Energieprofil für Excimer und Exciplex, das die Stärkung der AuI−AuI-Bindung entlang der Reaktionskoordinate aufzeigt.
Kristalle oder Pulver der Verbindung (DNP)[Au(CN)2]2⋅4 H2O (DNP2+=1,1′-Bis-(2,4-dinitrophenyl-4,4′-bipyridinium), in der die Anionen als Dimere zwischen den großen Kationen liegen, zeigen bei 10 K zwei Emissionsbanden im sichtbaren Bereich, die den Einheiten DMP2+ und {[Au(CN)2]2}2− zugeordnet werden können.116 Hier ist nur die letztgenannte Bande von Interesse. Die Anregung im nahen UV-Bereich und die Emission bei ca. 420 bzw. 580 nm sind charakteristisch für [Au(CN)2]−-Salze. Bei Temperaturen oberhalb von 50 K tritt aufgrund der Gegenwart des DNP2+-Kations schnelle Löschung durch Elektronenübertragung ein, ähnlich wie in der Reaktion zwischen 3[Au2(dppm)22+]* und MV2+ [Gl. (2)].163, 183
Fünf kürzlich erschienene Publikationen stützen nicht nur nahezu alle Ergebnisse aus dem Patterson-Labor zum Ursprung und zur Zuordnung der Emissionsspektren von konzentrierteren [Au(CN)2]−-Komplexlösungen und dem Typ der für die Lumineszenz-Eigenschaften verantwortlichen Excimer-/Exciplex-Bildung, sondern sie werfen auch ein neues Licht auf die nach der ursprünglichen Anregung stattfindende, ultraschnelle Strukturdynamik in derartig aktivierten chemischen Prozessen.
Die ultraschnelle Dynamik der Au−Au-Bindungsbildung im gestaffelten Dimer [Au(CN)2−]2 im angeregten Zustand wurde mittels zeitaufgelöster kurzlebiger Absorptionsspektroskopie (TRTA) im Femto- und Picosekunden-Bereich aufgeklärt. In sehr verdünnten [Au(CN)2]−-Lösungen (ca. 0.04 m) befinden sich hauptsächlich Dimere, die auch als einzige Spezies im nahen UV-Bereich (ca. 270 nm) angeregt werden. Für nachfolgende Prozesse wurden die Relaxationsübergänge wie auch die Zeitkonstanten bestimmt.109
Während der Photoanregung (Schema 9) spiegeln kohärente molekulare Bewegungen des im Femtosekunden-Bereich existierenden, angeregten Dimers im S1-Singulett-Zustand die schnelle Bildung einer starren kovalenten Au−Au-Bindung wider. Eine weitere Kontraktion der Au−Au-Bindung findet während des sich anschließenden Intersystem-Crossing in einen kurzlebigen Triplett-Zustand niedriger Energie des Dimers T1 (Zeitkonstante 0.21 ps) statt. Dieser stabile T1-Zustand zerfällt langsam (Emission bei 330 nm) mit einer Zeitkonstante von 26 ps. Im angeregten Zustand findet unter den gewählten experimentellen Bedingungen keine weitere Oligomerisierung statt.

Vereinfachtes Jablonski-Diagramm für die Anregung und Emission des Dimers [Au(CN)2−]2. (A=Absorption; IT=interner Übergang; ISC=Intersystem-Crossing; P=Phosphoreszenz).
Für die Oligomere [Au(CN)2−]n (n=2–5) wurden von Dolg, Thiel et al. Elektronenstruktur-Rechnungen auf dem MP2-Niveau (Grundzustand; erster angeregter Zustand des Dimers) und auf DFT, RICC′′ sowie MSCASPT2 (angeregte Zustände) durchgeführt.108 Die theoretischen Ergebnisse für das Dimer stehen in vollem Einklang mit den von Iwamura, Tahara und Mitarbeitern für die gestaffelte Struktur berichteten.110 Im Gegensatz zum Grundzustand, wo das ekliptische Isomer instabil ist, haben die Bindungskonfigurationen dσ*1pσ1 und dσ*1pπ1 des ekliptischen Dimers im angeregten Zustand für beide Zustände S1 und T1 recht ähnliche Energien und könnten theoretisch ebenfalls zu den Absorptions- und Emissionsbanden beitragen. Die berechneten Au−Au-Bindungslängen für die beiden Konformationen im aktivierten Singulett- und Triplett-Zustand sind nicht signifikant verschieden; sie sind beide ca. 0.3 Å kürzer als in den entsprechenden Grundzuständen. Nachdem die experimentellen Ergebnisse für den dimeren Komplex mit der vorgeschlagenen Strukturdynamik des gestaffelten Isomers in Einklang stehen, werden die für die ekliptische Form berechneten, ziemlich komplexen spektroskopischen Übergänge nicht weiter diskutiert.
Sich in den Fußstapfen von Patterson, Fackler und Mitarbeitern bewegend und den letzten experimentellen und theoretischen Charakterisierungsansätzen vertrauend haben sich andere Forschungsgruppen eingehender mit der Photophysik und Photochemie von [Au(CN)2−]n-Oligomeren in Lösung beschäftigt, insbesondere mit der des dreikernigen Komplexes (n=3). Iwamura, Tahara und Mitarbeiter109 haben die TRTA-Spektroskopie und auch zeitabhängige DFT-Berechnungen zur Erklärung der Strukturveränderungen während der photochemischen Emissionsübergänge im angeregten Zustand eingesetzt. Nachdem Sie die Anregungs- und Emissionsübergänge des trimeren [Au(CN)2−]3 sorgfältig von anderen störenden photochemischen Übergängen getrennt hatten, kamen sie zu dem Schluss, dass mindestens fünf verschiedene kinetische Ereignisse und fünf Komplexe zur vollständigen Erklärung der Absorptions- und Emissionsbanden nötig sind. Es wurden drei Zeitkonstanten für aufeinanderfolgende Übergänge bestimmt. Eine im Femtosekunden-Bereich identifizierte, entscheidende, kohärente Oszillationsbewegung gab einen starken Hinweis auf eine ultraschnelle Au−Au-Bindungsbildung im angeregten Zustand.
Alle vorgeschlagenen spektroskopischen Übergänge mit ihren Zeitkonstanten und begleitenden Strukturänderungen sind in den Schemata 10 und S1 (siehe Hintergrundinformationen) gezeigt. Zu beachten ist, dass das in Lösung vorliegende, schwach assoziierte Trimer [Au(CN)2−]3 bei einer gegebenen Konzentration und unter Umgebungsbedingungen eine gewinkelte (nicht-lineare) Struktur im Grundzustand zeigt, welche in der Franck-Condon-Region bei Anregung unverändert bleibt, allerdings augenblicklich in eine ähnliche, aber kovalent gebundene Struktur relaxiert. Während dieser Schwingungsrelaxation erfolgt scheinbar die Stärkung der Au−Au-Bindung (Stufe 1). Der angeregte Singulett-Zustand S1 ist fluoreszierend. Das Intersystem-Crossing (Stufe 2) unter Bildung von 3a[Au(CN)2−]3* ist innerhalb von 500 fs vollzogen. Die messbare zweite kinetische Komponente (Stufe 3) beinhaltet lediglich die Entfaltung des Au3-Gerüsts in die gestaffelte lineare Anordnung in 3b[Au(CN)2−]*. Danach wächst das Oligomer unter Beibehaltung der linearen Anordnung der Goldatome (Stufe 4). Der Triplett-Zustand niedrigster Energie verliert Energie durch Phosphoreszenz. Schnelle Energieänderungen aufgrund von Emission und numerische Zeitbeschränkungen wurden experimentell ermittelt, aber die Zuordnung zu verschiedenen Strukturänderungen sind hypothetisch und berechnet.

Jablonski-Diagramm für die spektroskopischen Energieniveaus des Trimers [Au(CN)2−]3109 (in blau); Strukturänderungen: 1) Verkürzung der Au-Au-Bindung, 2) ISC innerhalb von 500 fs, 3) Modifizierung der Struktur von gewinkelt nach linear im Triplett-Zustand innerhalb von 2.1 ps, 4) Assoziation von 3b[Au(CN)2−]3* (linear, Lebensdauer 2 ns) mit einem Monomer im Grundzustand unter Bildung eines linearen Tetramers. (A)–(C) (in rot, falls unterschiedlich) zeigen die später in den Referenzen 108 sowie 111–113 vorgeschlagenen Strukturveränderungen.
Die Emissions-Wellenlängen (Energien) und die Lebensdauern der [Au(CN)2]−-Oligomere in wässriger Lösung werden von der molekularen Umgebung im angeregten Zustand beeinflusst. Überraschenderweise zerfallen die [Au(CN)2−]n*-Oligomere (n≥4) durch Dissoziation in kleinere Excimere und Exciplexe, es konnte aber gezeigt werden, dass Wechselwirkungen mit Tetraalkylammonium-Kationen wie Et4N+ die größeren Oligomere stabilisieren und die Emissionen niedrigerer Energie verstärken.222
Photoanregung von Dicyanidoaurat(I)-Anionen in ionischen Flüssigkeiten mit 1-Butyl-3-methylimidazolium- und N-Butyl-N-methylpyrrolidinium-Kationen im nahen UV führt zu einer Fluoreszenz-Bande bei ca. 380 nm und zu Phosphoreszenz bei 460 nm. Die Position der letzten Bande wird mit der Zeit zu niedrigerer Energie verschoben. Es wurden zwei Zeitkonstanten für jede ionische Flüssigkeit, jeweils für die Triplett-Bildung und die Relaxation, bestimmt. Die Autoren dieses Berichts114 beriefen sich in der Erklärung ihrer Daten auf die von Iwamura und Mitarbeitern für die trimere Spezies [Au(CN)2−]3* (Schema 10) vorgeschlagene Dynamik. Wieder wurden zwei Struktursituationen für die Triplett-Trimere berücksichtigt, obwohl es experimentelle Hinweise für nur eine gab. Die Verschiebung der Bande zu längeren Wellenlängen wurde über die von der Größe des Kations beeinflusste Bildung höherer [Au(CN)2−]n*-Oligomere interpretiert.
Die Interpretation der im aktivierten Zustand während der Anregung von Lösungen von [Au(CN)2]− auftretenden, progressiven Veränderungen durch Iwamura, Tahara und Mitarbeiter wurde in kürzlich erschienen Artikeln herausgefordert. Darin stimmen die Autoren generell mit den vorgeschlagenen Abläufen überein, aber sie unterscheiden sich in Bezug auf die genaue Kinetik der frühen Übergänge.
Auf demselben Theorie-Niveau, das für die Interpretation der photophysikalischen Vorgänge im [Au(CN)2]−-Dimer (siehe oben) herangezogen wurde, kamen Fang, Dolg, Thiel und Mitarbeiter108 zu dem Schluss, dass die gestaffelte gewinkelte Struktur für das Trimer in den Schwingungs-Grundzuständen der Singulett- oder angeregten Triplett-Zustände nicht existiert. Folglich wird die lineare Anordnung bereits schnell während der Schwingungsrelaxation im aktivierten Singulett-Zustand eingenommen und von der Au⋅⋅⋅Au⋅⋅⋅Au-Bindungsverkürzung begleitet [(A) in Schema 10]. Anschließend führt das Intersystem-Crossing [(B) in Schema 10] für das Trimer zu einem Triplett-Zustand niedriger Energie, welcher den endgültigen linearen Strukturen in Schema 10 und S1 entspricht.
Motiviert durch diese im vorherigen Abschnitt beschriebenen, gegensätzlichen Ergebnisse haben Kim et al.111, 112 die erste zeitaufgelöste Röntgenstreuung (TRXSS) an gelösten Goldkomplexen durchgeführt. Sie haben sich explizit auf das photoinduzierte dynamische Verhalten des [Au(CN)2]−-Komplexsystems während und nach der Anregung fokussiert unter Einsatz eines Freie-Elektronen-Röntgen-Lasers zur Untersuchung ultraschneller Reaktionen. Der große Vorteil von TRXSS gegenüber der TRTA-Methode liegt in ihrer Empfindlichkeit gegenüber der globalen Molekülstruktur in Lösung. Die Technik ist somit gut geeignet für die direkte Charakterisierung individuell vorherrschender, kurzlebiger Komplexspezies mit einer räumlichen Auflösung im sub-Å-Bereich. Mit einer zeitlichen Auflösung im sub-ps-Bereich können Informationen zur Bindungsverkürzung, Bildung der kovalenten Bindung, Transformation von gewinkelt nach linear und zum Oligomer-Wachstum entlang einer gewählten Reaktionsachse geliefert werden.
Die Ergebnisse von Kim et al. stimmen völlig mit den früher auf der Basis quantenchemischer Berechnungen108 erhaltenen überein. Im Gegensatz zu den Resultaten von Iwamura, Tahara und Mitarbeitern wurde aber nun der folgende Relaxationsweg formuliert (vergleiche Schema 10 und S1): A) die Anregung bewirkt augenblicklich (innerhalb von 500 fs) die Umlagerung von gewinkelt nach linear in der Franck-Condon-Region; B) innerhalb von 1.6 ps wird durch S1→T1 (Trimer)-ISC die einzige involvierte Triplett-Situation erreicht, bevor intermolekulare Wechselwirkung eintritt, und eine weitere Stärkung der Au−Au−Au-Bindungen stattfindet; C) die kinetische Zeitkonstante für den spektroskopischen T1 (Trimer)→T1 (Tetramer)-Übergang durch Au-Au-Atomorbital-Überlappung beträgt 1.3 ns; anschließend (D) stehen dann noch 100 ns für die vollständige Relaxation in den Grundzustand, T1 (Tetramer)→S0, zur Verfügung.
Veranlasst durch den Einwand von Iwamura, Tahara und Mitarbeitern,110 dass die Diskrepanzen zwischen den TRTA- und TRSXX-Ergebnissen daran liegen könnten, dass Kim et al. nicht die für eine selektive Anregung des Trimers optimale Wellenlänge benutzt hätten, hat die letztgenannte Gruppe alle Messungen unter Verwendung von zwei verschiedenen Lösungen bei niedrigerer Anregungsenergie (jetzt 310 nm, vorher 287 nm) wiederholt. In überzeugender Manier konnten dieselben Rückschlüsse gezogen werden wie zuvor, in den Worten von Kim et al.: “der Übergang von gewinkelt nach linear des [Au(CN)2−]3-Trimers erfolgt innerhalb von 500 fs” und “der Mechanismus der Au-Au-Bindungsbildung im [Au(CN)2−]3-Trimer ist unabhängig von der Anregungs-Wellenlänge”.113 Es sollte erwähnt werden, dass die experimentell ermittelte Au-Au-Bindungsverkürzung nur 0.11 Å beträgt, mit einer ziemlich hohen Standardabweichung von ca. 0.04 Å.
In ihrem jüngsten Beitrag haben Tahara und Mitarbeiter223 ihre TRTA-Ergebnisse durch Verfolgen der ultraschnellen Strukturdynamik von [Au(CN)2−]3 während der Au−Au-Bindungsbildung mittels Time-Domain-Raman-Tracking ergänzt. Die Au−Au-Atmungsschwingung wurde bei ca. 90 cm−1 gemessen. Der anfänglichen Zunahme der Raman-Intensität aufgrund von Au−Au-Bindungsbildung und Intersystem-Crossing folgt innerhalb weniger Picosekunden eine graduelle Hochfeldverschiebung der Frequenz, was eine kontinuierliche, strukturelle Veränderung wie den vorgeschlagenen Übergang von gewinkelt nach linear auf der Potentialfläche des Triplett-Zustands stützt. Tahara und Mitarbeiter haben auch aufgezeigt, dass die Ergebnisse von Time-Domain-Raman-Messungen selbst unter den für die Triplett-Anregung nahezu idealen Bedingungen aufgrund der gleichzeitig stattfindenden Anregung des Tetramers aus dem Grundzustand und der nachfolgenden Relaxation in einen Triplett-Zustand verkompliziert werden. Die Gegenwart dieses angeregten Tetramers kann auch für die oben beschriebenen widersprüchlichen Röntgenstreuungsergebnisse in Lösung mitverantwortlich sein.108, 111, 112
4.3.2 Der Fall Di(thiocyanato)aurat(I)
Die wegweisenden Ergebnisse von Patterson, Fackler und Mitarbeitern zur Excimer- und Exciplex-Bildung von [Au(CN2)]−-Komplexen in Lösung und in dotierten Kristallen haben zu Untersuchungen der Lumineszenz von M[Au(SCN)2]-Komplexen im Festkörper und in gefrorenen Lösungen inspiriert.140, 142 Als Alleinstellungsmerkmal werden für eine Reihe von Gegenionen M+=K+, Rb+, nBu4N+ und Cs+ bei Raumtemperatur und bei 77 K sowohl blaue Fluoreszenz- als auch grüne, unstrukturierte Phosphoreszenz-Banden beobachtet. Die Hauptemissionsbande ist in gefrorenen Lösungen rotverschoben gegenüber dem kristallinen Material. Eine Verschiebung zu niedrigeren Energien derselben Bande wird auch mit zunehmender Größe des gewählten Gegenions beobachtet. Insbesondere Ph4P+-Salze, in denen die Au(SCN)2]−-Einheiten im Festkörper getrennt voneinander vorliegen, zeigen keine Au-zentrierten Emissionen. Gleiches wurde zuvor für das Cyanid-Analogon nBu4N[Au(CN)2] berichtet.107
Die Emission bei hoher Energie entspricht einem emittierenden angeregten Singulett-Zustand (<500 nm), und die andere Bande (>500 nm) stammt von einem T→S0-Übergang. Die Molekülstrukturen der lumineszierenden Zustände und des Grundzustands wurden mittels quantenchemischer Berechnungen auf MP2- und DFT-Niveau unter Verwendung des [Au(SCN)2−]2-Dimers (Formel K, siehe oben) als Modellsystem ermittelt. Diese dimere Einheit ist realistisch, da sie in kristallinem nBu4N[Au(SCN)2] vorkommt. Es wird vorhergesagt, dass die Bildung der Au−Au-Excimer-Bindung im angeregten Zustand mit einer deutlichen Verkürzung der Bindungslänge von 0.2–0.3 Å (abhängig vom verwendeten Theorie-Niveau) einhergeht. Auf der Basis von Rechnungen für Aggregate in der Gasphase, die eine zu große Rotverschiebung ergeben, wenn das Dimer weiter oligomerisiert, haben die Autoren angenommen, dass hauptsächlich diskrete zweikernige Excimer-Einheiten für die beobachtete Lumineszenz verantwortlich sind. Solche angeregten Dimere [Au2(SCN)42−]* liegen dann in Kristallen der verschiedenen Salze vor, unabhängig davon, ob die Anionen im Grundzustand der ausgedehnten Strukturen dieser Salze als Dimere, Trimere oder als Ketten angeordnet sind. Es wurde festgestellt, dass die Größe des Kations einen Einfluss auf die Erscheinungsform der Spektren hat. Die Beteiligung der Gegenionen hängt mit unspezifischen Verzerrungen zusammen und nicht mit einer Exciplex-Bildung, da die größeren Kationen hinsichtlich ihres bathochromen Effekts einen größeren Einfluss ausüben. Je größer das Gegenion ist, umso weniger Beschränkungen gibt es für den photoinduzierten Strukturwechsel im angeregten Zustand. Gefrorene Lösungen gestatten mehr Freiheiten als Festkörper.
Einer “Dimer-Hypothese” wurde in einer anderen Untersuchung an neutralen Komplexen des Grundtyps [(RNC)AuX] widersprochen. Die Zunahme der Verzerrung im angeregten Zustand und die Abnahme der Emissionsenergie mit dem Schrumpfen der Au−Au-Bindung “erfolgt über einen längeren Bereich als den im Dimer”.224 Balch und Mitarbeiter225 haben bei ihren Untersuchungen an neutralen lumineszierenden (Cyclohexylisocyanid)gold(I)-Halogeniden mit relativ schwachen aurophilen Wechselwirkungen im festen Zustand auch gefolgert, dass “die spektroskopischen Eigenschaften […] eine Konsequenz der supramolekularen Organisation von Molekülen innerhalb der Festkörper sind”. Exciplex-Bildung und strukturelle Verzerrung bleiben in all diesen Komplexen dennoch wichtige Merkmale der Dynamik im angeregten Zustand (vergleiche Abschnitt 4.3.4 unten).
4.3.3 Die Fälle Mono- und Bis(phosphin)gold(I)
Die Lumineszenz niedriger Energie (λmax>500 nm) bei 70 K von kristallinem [(Et3P)AuCl] wurde Gold-zentrierten Übergängen zugeschrieben. DFT- und ZINDO-Rechnungen stützen diese Annahme. Die Lumineszenz wird in Lösung vollständig gelöscht.226 Strukturelle Informationen zu diesen Komplexen in den angeregten Zuständen liegen nicht vor.
Die Emissions-Spektren der homoleptischen, über aurophile Wechselwirkungen assoziierten kationischen Komplexe [(Me3NBH2MH2)2Au]AuCl4 (M=P, As) wurden im Festkörper untersucht.146 In den Kristallen bilden die Komplexe Ketten, und die beiden Emissionen jedes einzelnen Komplexes im blauen bis violetten Bereich sind (laut DFT-Rechnungen) MC und finden zwischen Leitungs- und Valenzband-Konfigurationen statt. Die Energie des Intersystem-Crossing T1→S0 (letzte Relaxation) könnte manipulierbar sein: Absenken der Temperatur führt zu einer Verkürzung des Au-Au-Abstands im Grundzustand, die Orbitalüberlappung im angeregten Zustand nimmt zu, die Bandlücke wird kleiner, und es resultiert eine Rotverschiebung. Der Einfluss der Temperatur auf die Au-Au-Abstände in linearen Ketten von Goldkomplexen in Festkörpern spiegelt gewissermaßen die früher in diesem Artikel diskutierte (Abschnitt 4.3.1) Erhöhung des hydrostatischen Drucks wider.
Trotz bemerkenswerter Fortschritte, die beim Verständnis der Photophysik und den chemischen Umlagerungen der Excimere und Exciplexe von durch aurophile Kontakte präorganisierten, einkernigen Gold(I)-Komplexen gemacht worden sind, gibt es noch eine Reihe von ungeklärten Sachverhalten, die weitere Aufmerksamkeit verdienen. Darunter befindet sich die Rolle der Kationgröße und -ladung für die Strukturfestlegung und die Energie der angeregten Zustände und deren Lebensdauer in Festkörpern und ionischen Flüssigkeiten, die Excimer-Eigenschaften in spezifischen Positionen einer festen Matrix und in ein- und zweidimensionalen Anordnungen von Gold(I)-Zentren, sowie Details der Relaxationsprofile von [Au(CN)2−]n* und [Au(SCN)2−]n* (n≥4) in Lösung.
5 Die Rolle zweikerniger Gold(I)-Excimere und -Exciplexe in der organischen Synthese
Die bahnbrechenden Untersuchungen von Che und Mitarbeitern zum Einsatz von photo-aktivierten, Phosphin-verbrückten zweikernigen Gold(I)-Komplexen (N) als Oxidations- oder Reduktionsmittel und schließlich als Katalysatoren für die C-C-Bindungsknüpfung186 haben in letzter Zeit andere Forschungsgruppen dazu animiert, solche schwach Au⋅⋅⋅Au-gebundenen Komplexe als präkatalytische Mediatoren in der Photokatalyse einzusetzen. Der Terminus “photoredox binuclear gold catalysis” umspannt inzwischen einen weiten Bereich von Anwendungen in der präparativen organischen Chemie. Wichtige Beiträge wurden unabhängig von den Forschungsgruppen von Barriault, Hashmi und anderen geleistet. Die wichtigsten Ergebnisse sind schon von Barriault und Mitarbeitern kurz und klar zusammengefasst worden227, 228 und werden hier nicht im Detail wiederholt. Eine Vielzahl von Reaktionen wie C-X-Bindungsbruch, C-C- und C-N-Kreuzkupplung, Pinakol-Umlagerung und selbst Heck-ähnliche Additionen wurden erfolgreich katalysiert. In den entsprechenden Katalysezyklen wird vorgeschlagen, dass das in einem langlebigen Triplett-Zustand existierende [AuI−AuI]*-Excimer während der Bestrahlung der Reaktionsmischung mit abstimmbaren Lichtquellen als aktives Zentrum erscheint. Anwendungen in Totalsynthesen (±-Triptolid in der Gruppe von Barriault;229 Pyrroloazocinindol-Alkaloide von Echavarren und Mitarbeitern230) und radikalischen Polymerisationen (von Nzulu et al.)231 haben Potential und Anwendungsbreite des Ansatzes überzeugend aufgezeigt. Von Hashmi et al. wurde mit dem Einsatz von Co-Reduktionsmitteln wie tertiären Phosphinen oder Thiolen eine sehr vielversprechende Variante gefunden.232 Diese Publikation und andere Berichte aus jüngster Zeit233 enthalten Schlüsselreferenzen, die eine Verfolgung der aktuellen Aktivitäten gestatten.
6 Zusammenfassung und Ausblick
In den letzten dreißig Jahren wurde die Goldchemie als ein Forschungsgebiet erkannt, in dem für eine Unmenge von Verbindungen sowohl im Festkörper als auch in Lösung die Bildung und Relaxation von Excimeren und Exciplexen leicht zu beobachten sind. Diese weite Verbreitung liegt an der spontanen seitlichen Aggregation von linear zweifach koordinierten Gold(I)-Komplexen über aurophile Wechselwirkungen. In diesen Aggregaten werden die Goldatome im engen Kontakt (mit Au-Au-Abständen von ca. 3.0 Å) gehalten. Bei UV/Vis-Anregung werden diese Au⋅⋅⋅Au-Kontakte signifikant verkürzt, was die Bildung kovalenter Au−Au-Bindungen im angeregten Zustand anzeigt. Sowohl die für diese neuartigen [Au−Au]*-Bindungen gefundenen und berechneten Bindungslängen als auch die Bindungsenergien sind mit denen der Au-Au-Bindung im Goldmolekül Au2 und seinen Komplexen L−Au−Au−L ebenso wie mit denen in zweikernigen Gold(II)-Komplexen vergleichbar.
Typischerweise handelt es sich bei der UV/Vis-Anregung um einen Metall-zentrierten Prozess [Au2]→[Au2]*, der ohne direkte Beteiligung der Liganden zu einer Reihe von angeregten Singulett- und Triplett-Zuständen führt. Generell äußert sich die Relaxation quantitativ in Lumineszenz-Effekten, die die Vorgänge unmittelbar sichtbar machen und detaillierte quantitative Untersuchungen zur Kinetik durch zeitabhängige Experimente gestatten. Die Anregung ist mit signifikanten Strukturveränderungen in Bezug auf die Bindung der Metallatome verbunden. Wenn bei der Anregung elektronische Zustände der Liganden involviert werden, d. h. wenn der Prozess nicht strikt Metall-zentriert abläuft, sind vergleichbare Auswirkungen auf die Au-Au-Bindungen hingegen weniger ausgeprägt oder gar nicht vorhanden.
Besonders detaillierte Informationen über die Energieprofile der Excimere und Exciplexe wurden für den Dicyanidoaurat(I)-Komplex [Au(CN)2]− zusammengetragen, der sowohl im Festkörper als auch in Lösung über aurophile Kontakte assoziiert ist. In Abhängigkeit von den Konzentrationen in Lösungen oder von der Dotierungskonzentration in Festkörpern können die Dimere, Trimere und Tetramere beobachtet, und ihre Umwandlung in Excimere und Exciplexe über zeitaufgelöste Picosekunden/Femtosekunden-Emissions- und -Absorptionsspektroskopie sowie Femtosekunden- Röntgenstreuung in Lösung verfolgt werden. Als weiteres spektroskopisches Werkzeug wurde zur Charakterisierung der Au−Au-Bindung im [(NC)2Au−Au(CN)22−]*-Excimer die Resonanz-Raman-Spektroskopie eingesetzt.
Die experimentellen Untersuchungen wurden durch umfangreiche quantenchemische Berechnungen ergänzt, die einen tieferen Einblick in die strukturellen Eigenschaften und die photoinduzierte Dynamik der angeregten Zustände gaben. Die Untersuchungen des Di(thiocyanato)gold(I)-Komplexes [Au(SCN)2]− ergaben ähnliche Ergebnisse, was zeigt, dass die Eigenschaften für kleine Gold(I)-Komplexe generelle Gültigkeit haben.
Das zweite Gebiet, in dem Gold(I)-Excimere häufig angetroffen werden, sind zweikernige Gold(I)-Komplexe mit einer cyclischen Struktur, in denen der intramolekulare aurophile Kontakt durch verbrückende Liganden gestützt wird. Bei UV/Vis-Anregung verkürzt sich der transannulare Au⋅⋅⋅Au-Kontakt beispielsweise in einer achtgliedrigen dimetallacyclischen Verbindung unter Bildung einer bicyclischen Struktur, in der die neu generierte Au−Au-Bindung zwei fünfgliedrigen Ringen gemeinsam ist. Die Excimere wurden wieder über UV/Vis-Absorptions- und -Emissions-Spektroskopie, Raman-Untersuchungen und schließlich über elektrochemische Studien, in denen die Veränderungen der Redox-Potentiale abgeschätzt wurden, charakterisiert. Der Einfluss der Natur der verbrückenden Liganden, Gegenionen und Lösungsmittel ist bei den Dimetallacyclen häufiger zu verfolgen, da sie in einer Reihe von Lösungsmitteln untersucht werden können, welche an den Anregungs- und Emissions-Prozessen und an der Bildung von Exciplexen beteiligt sein können. Für Komplexe mit typischen Bis(phosphin)-Liganden R2PCH2PR2 ermöglichte jüngst eine ultraschnelle, zeitaufgelöste transiente Absorptions- und Emissions-spektroskopische Untersuchung die Identifizierung des intrinsischen Excimers im Triplett-Zustand (mit einer Au−Au-Bindung), welches unmittelbar bei Bestrahlung als das Produkt eines mit ultraschneller Geschwindigkeit stattfindenden, und ohne Beteiligung von Phase und von Lösungsmittel-Molekülen ablaufenden Prozesses gebildet wird. Nachfolgende Wechselwirkungen können dann zu Exciplexen mit anderen Monomeren, Solvens-Molekülen oder Gegenionen führen. Interessanterweise zeigen die Goldatome in den Exciplexen einen stärkeren elektrophilen Charakter und akzeptieren leichter Donor-Moleküle oder Anionen als solche im Grundzustand, in dem die Metallzentren streng zweifach koordiniert sind. Cyclische [Au2(P^P)2]2+-Komplexe ergeben Excimere, welche Schlüsselintermediate in der in der organischen Synthese angewandten “photoredox binuclear gold cataysis” sind.
Die Rückschlüsse aus den in experimentellen Untersuchungen gesammelten Daten wurden auch in quantenchemischen Berechnungen bestätigt, welche eine Abschätzung der Eigenschaften der kurzlebigen Intermediate und Übergangszustände erlaubt haben. In mehrkernigen Verbindungen mit Goldatomen, die über multifunktionale Liganden mit unterschiedlichen Donor-Atomen in Ketten angeordnet sind, kann das photophysikalische Verhalten ihrer Excitonen über die Wahl der Ligandeigenschaften eingestellt werden.
Ausgewählte Beispiele von Silber(I)-Analoga wurden ebenfalls bezüglich einer Excimer-Bildung getestet, und für nahezu alle von ihnen wurden ähnliche Anregungsprozesse bestätigt. Obwohl argentophile Wechselwirkungen in den Vorstufen-Aggregaten generell als schwächer eingestuft werden als in den Gold(I)-Analoga, wurde in den Excimeren sogar eine stärkere Ag−Ag-Bindung gefunden, vermutlich deshalb, weil Silber(I) höhere Koordinationszahlen bevorzugt, was die Bindung in Exciplexen mit drei- oder vierfach koordinierten Metallatomen unterstützt. Diese Beobachtung zeigt, dass starke relativistische Effekte die metallophile Bindung im Grundzustand von Gold(I) stärker unterstützen als von Silber(I), dass diese aber für die angeregten Zustände der beiden Münzmetalle weniger bedeutend sind. Es gibt immer noch wenige Informationen zu Excimeren in der Quecksilber- oder Thallium-Chemie, wo metallophile Bindungen bisher nur eine untergeordnete Rolle spielen. Wie oben erwähnt, wurden die ersten Beispiele für eine Excimer-Bildung aus Metallkomplexen an Beispielen mit Platin(II) gefunden, wo seitliche Pt⋅⋅⋅Pt-Kontakte ausgebildet werden, durch die ein Paar aus quadratisch-planar koordinierten Metallatomen entsteht, welches dann als Vorstufe für die Excimer-Bildung fungiert.
Acknowledgements
Die Autoren danken der Alexander-von-Humboldt-Stiftung für die großzügige Unterstützung.
Conflict of interest
Die Autoren erklären, dass keine Interessenkonflikte vorliegen.
Biographical Information
Hubert Schmidbaur ist emeritierter Professor an der TU München. Nach dem Chemiestudium an der LMU München war er an den Universitäten Marburg und Würzburg sowie mehrfach im Ausland tätig. Nach Arbeiten auf sehr verschiedenen Gebieten wie der Silicium- und Phosphorchemie, den π-Komplexen von Hauptgruppenelementen sowie der bioanorganischen Chemie von Magnesium und anderen Elementen lag sein Forschungsschwerpunkt über lange Jahre auf der Chemie von Gold, für die er mehrfach ausgezeichnet wurde.

Biographical Information
Professor Helgard Raubenheimer ist emeritierter Professor an der University of Stellenbosch (SUN), Südafrika. Er studierte Chemie an der SUN und war Professor an der Rand Africaans University Johannesburg, bevor er zur SUN als Direktor des Department of Chemistry and Polymer Sciences zurückkehrte. Er hat Sabbaticals mit E. O. Fischer und H. Schmidbaur (TU München) sowie D. Seebach (ETH Zürich) verbracht. Seine Forschung behandelt Carbenkomplexe von Übergangsmetallen, mit einem Fokus auf Goldchemie in den letzten Jahren, wofür er viele Auszeichnungen in Südafrika und im Ausland erhalten hat.


