

2-Hydroxybenzophenone as a Chemical Auxiliary for the Activation of Ketiminoesters for Highly Enantioselective Addition to Nitroalkenes under Bifunctional Catalysis
Abstract
An organocatalytic system is presented for the Michael addition of monoactivated glycine ketimine ylides with a bifunctional catalyst. The ketimine bears an ortho hydroxy group, which increases the acidity of the methylene hydrogen atoms and enhances the reactivity, thus allowing the synthesis of a large variety of α,γ-diamino acid derivatives with excellent stereoselectivity.
α-Amino acids are essential molecules in many fields. They are used in the synthesis of peptides and proteins, as chiral catalysts, as a source of chirality in the design of ligands, and in total synthesis.1 Such is the demand for enantiomerically enriched α-amino acids that synthetic organic chemists continue to develop new methods for their synthesis.2 Especially important and relevant are α,γ-diamino acid derivatives, which are present in a large number of natural and pharmaceutical products.3 For example, DABA analogues are used as a variety of drugs, such as anticonvulsants, sedatives, and anxiolytics,3a HA-966 and L687414 are used as NMDA antagonists,3b,3c and cucurbitine, a natural product found in pumpkins, is used against the parasite Schistosoma japonicum3d (Scheme 1, middle right).

Background and strategy for the synthesis of a novel chemical auxiliary in the organocatalytic synthesis of α,γ-diamino acid derivatives. Boc=tert-butoxycarbonyl.
Ketiminoesters derived from glycine have become increasingly important because they provide a starting material for the synthesis of optically pure α-amino acid derivatives.4 The alkylation of glycine ketimines with different alkylating reagents under phase-transfer catalytic conditions has been well developed by the research groups of O'Donnell,5 Maruoka,6 and others.7 However, examples of their addition to nitroalkenes, which would give access to α,γ-diamino acid derivatives, are scarce (Scheme 1, middle).8, 9 No examples of highly enantioselective organocatalytic addition reactions of monoactivated ketimines to nitroalkenes have been reported.9b The reason for this absence of reactivity, especially in the field of bifunctional thiourea catalysis,9a,9b is related to the lack of acidity of the hydrogen atoms of the α-methylene ester. In fact, for this class of ylides, two electron-withdrawing groups (EWGs) are needed to increase the acidity and promote the organocatalytic addition.
During our investigations on bifunctional catalysis,10 we speculated that it might be possible to overcome this organocatalytic limitation in the synthesis of α,γ-diamino acid derivatives. Recently, different research groups, such as those of Takemoto,11 Vicario,12 Palomo,13 and Krische,14 among others15 (Scheme 1, top), have shown how different chemical auxiliaries can increase the electrophilicity of amides or ketones via hydrogen-bonding activation, or be used to control stereoselectivity and reactivity (nitroalkenes and aldehydes). However, only a few studies have been reported in which this approach has been used to increase nucleophilicity.
The low reactivity of azomethine ylides in organocatalytic reactions is due to the low acidity of the CH2 hydrogen atoms. Therefore, we wondered whether activation by intramolecular hydrogen bonding would help in the addition to nitroalkenes and provide good reactivity and enantioselectivity. However, for the development of an appropriate scaffold, the chemical auxiliary must be recyclable, inexpensive, and readily removable. Taking these factors into consideration, we hypothesized that 2-hydroxybenzophenone may be a suitable candidate owing to the ready formation of a six-membered ring through intramolecular hydrogen bonding and the ready ketimine hydrolysis (Scheme 1, bottom). We describe herein a new direct process for the synthesis of γ,α-diamino acid derivatives by the use of a new chemical auxiliary that is readily recovered, activates the glycine ketimine, and provides excellent yields, diastereoselectivity, and enantioselectivity.
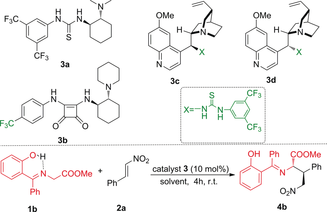
Initially, we synthesized ketimines 1 a and 1 b (Scheme 2) in good yields from commercially available starting materials (see the Supporting Information). The stability of ketimine 1 b is very high, and it could be stored at room temperature for months. As a proof of concept, we investigated the reaction of ketimine 1 a, without a hydroxy group, and ketimine 1 b with nitroalkene 2 a in the presence of the Takemoto catalyst 3 a. No reaction took place when ketimine 1 a was used, and after 24 h no trace of the corresponding product 4 a was detected. However, when ketimine 1 b was used with catalyst 3 a, a complete conversion into 4 b occurred in only 4 h. These preliminary results prompted us to optimize the reaction conditions in terms of enantioselectivity, diastereoselectivity, and yield (Table 1).

Proof of concept for the intramolecular H-bond activation.
Entry |
Cat. |
Solvent |
Conversion[b] |
dr[b] |
ee [%][c] |
|---|---|---|---|---|---|
1 |
3 a |
CH2Cl2 |
100 |
90:10 |
97 |
2 |
3 b |
CH2Cl2 |
10 |
80:20 |
n.d.[d] |
3 |
3 c |
CH2Cl2 |
89 |
92:8 |
95 |
4 |
3 d |
CH2Cl2 |
100 |
95:5 |
97 |
5 |
3 a |
p-xylene |
92 |
92:8 |
95 |
6 |
3 a |
DCE |
100 |
88:12 |
96 |
7 |
3 a |
Et2O |
84 |
85:15 |
96 |
8 |
3 a |
THF |
83 (100)[e] |
>98:2 |
98 |
9 |
3 a |
CH3CN |
94 |
90:10 |
96 |
10 |
3 a |
MeOH |
52 |
91:9 |
74 |
- [a] Reaction conditions: 1 b (0.2 mmol), 2 a (0.24 mmol), 3 (10 mol %), indicated solvent (0.4 mL). [b] Conversion and the diastereomeric ratio were determined by 1H NMR analysis. [c] The ee value was determined by supercritical fluid chromatography (SFC). [d] Not determined. [e] In brackets conversion after a reaction time of 15 h. DCE=1,2-dichloroethane.
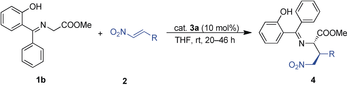
Different thiourea and squaramide catalysts 3 a,b and cinchona thioureas 3 c,d were tested (Table 1, entries 1–4). Similar results were found with all the thiourea catalysts (>95 % ee; entries 1 and 3–4), but very low conversion was observed with the squaramide catalyst 3 b (entry 2). We then continued the optimization with the commercially available Takemoto catalyst 3 a. Different apolar and polar solvents, such as 1,2-dichloroethane (DCE), Et2O, THF, CH3CN and p-xylene, produced similar results (entries 5–9), whereas protic solvent MeOH produced worse result (entry 10). Interestingly, ethereal solvents provided good results, especially in the case of THF, the use of which resulted in product 4 b as a single diastereoisomer with 98 % ee (entries 7 and 8). Conversion in the solvent THF was 83 % after 4 h, and was complete after 15 h (Table 1, entry 8). With these conditions defined, the scope of the reaction was studied with different substituted nitroalkenes (Table 2) and ketimines (Scheme 3).
|
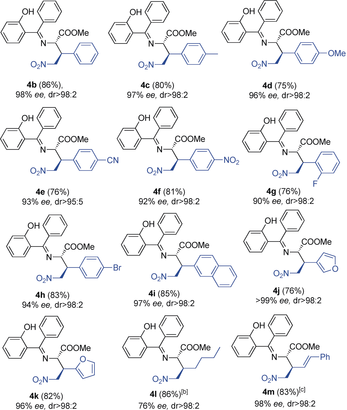
- [a] Reactions were performed on a 0.2 mmol scale in THF (0.4 mL); ee values were determined by SFC; diastereomeric ratios were determined by 1H NMR spectroscopy; yields are for the isolated product after flash chromatography. [b] Reaction time: 4 days. [c] Reaction time: 5 days.
When the nitroalkene contained an aromatic substituent with an electron-donating or an electron-withdrawing group, the reaction proceeded with high enantioselectivity (products 4 c–f, 92–97 % ee) in good yield (74–81 %). When a halogen substituent was in the ortho (product 4 g) or para position at the aromatic ring (product 4 h), or when a bulkier group, such as naphthyl (product 4 i) or a heterocycle (products 4 j and 4 k), was present instead of a phenyl group, the products were formed with good to excellent enantioselectivity (90–99 % ee). Interestingly, an alkyl group at the nitroalkene was also tolerated, yielding the amino acid derivative 4 l, which is difficult to obtain by other methods, but a longer reaction times of 4 days was needed. A substrate with a double bond in conjugation with the nitroalkene moiety led to the final product 4 m in good yield (83 %) with excellent enantioselectivity (98 % ee).
We analyzed other groups at the azomethine ylides in place of the carboxylate group (Scheme 3). This catalytic system with the hydroxy group allowed the addition of nitrile ylide 1 n and yielded the product 4 n with moderate enantioselectivity. However, the use of derivatives 1 o and 1 p, with a ketone and an amide, respectively, resulted in better enantioselectivity (93 % ee in both cases). The presence of CF3 or an aryl group did not enable the synthesis of the corresponding products 4 q and 4 r, and we only observed the unaltered starting materials in the crude mixture.

Use of different glycine ketimines. Reaction conditions: 1 (0.2 mmol), 2 (0.24 mmol), 3 a (10 mol %), THF (0.4 mL). [a] Yield based on recovered material.
When the reaction was scaled up, the hydroxyketone and catalyst 3 a were readily recovered (Scheme 4). The reaction on a 5.6 mmol scale took 22 h to reach completion. Upon fast percolation of the crude mixture, compound 4 b was obtained with 98 % ee, and catalyst 3 a was recovered in 75 % yield (Scheme 4, right). Compound 4 b was treated with HCl (10 %), and after a simple extraction, the hydroxyketone 5 was recovered in 96 % yield. The amino acid salt derivative 6 was washed with NaHCO3, and the free amino acid 7 was obtained (Scheme 4, left). The absolute configuration of 6 was unequivocally assigned as 2S,3S by X-ray crystallographic analysis (Scheme 4, top left), and the other reactions to give compounds 4 were assumed to have the same stereochemical outcome.

Scale-up and catalyst and chemical-auxiliary recovery.
We carried out DFT calculations to study the mechanism of the reaction of nitroalkene 2 a with the glycine ketimine 1 b in presence of catalyst 3 a (see the Supporting Information for details).17 Initially, we investigated the reasons for the lack of reactivity of the three nucleophiles 1 a, 1 q, and 1 r in comparison with 1 b and 1 n–p (Figure 1). We considered the acid–base equilibrium in which a proton is transferred from the ketimine 1 to the catalyst 3 a and a molecular hydrogen-bonded species 4A-complex is formed.18 The relative Gibbs free energy (ΔG) in the proton-transfer equilibrium is presented in Figure 1. We then evaluated the effect of different substituents on the acidity of ketimine 1 towards the formation of the ylide 4A-complex. The ΔG value for 1 b (with an OH group) is 5.9 kcal mol−1 lower than that for 1 a (without an OH group), which demonstrates the importance of the intramolecular H bond in increasing the acidity. Electron-withdrawing substituents, such as COPh (substrate 1 o), CN (substrate 1 n), CONMe2 (substrate 1 p), CO2Me (substrate 1 b) present both inductive (−I) and mesomeric (−M) effects; however, 1 q (CF3) possesses only an inductive (−I) and 1 r (Ph) exclusively a mesomeric (−M) effect. As was predicted, the reactants that contain groups with both effects showed a lower ΔG value (higher acidity). For groups at the ketimine 1 showing only one of the effects, the ΔG value was increased (lower acidity). Consequently, substrates 1 q and 1 r did not react. Therefore, a combination of these factors is needed to increase the acidity: the intramolecular hydrogen bond (C=N⋅⋅⋅HO−Ar) and appropriate substituents on the ketimine 1 (R groups with both −I and −M effects).

Relative Gibbs free energy of proton-transfer reactions with different nucleophiles 1.
We then considered the complete picture of the mechanism for the reaction between 1 b and 2 a catalyzed by 3 a. Two steps are involved in this process: proton transfer leading to the ylide, followed by C−C bond formation (Figure 2, top). The energetic profile of both steps is presented in Figure 2. In the first stage, there is a transition state for the direct migration of the proton from ketimine 1 b to the catalyst 3 a. The relative position of the nitroalkene 2 a depends on the relative position of the proton, because the intrinsic dipole of the nitro group points towards the positive charge on the proton and assists the proton migration. Figure 2 also shows how the coordination by means of hydrogen bonds is fundamental to the appropriate orientation of ketimine 1 b with respect to the catalyst 3 a, enabling the proton transfer to occur. The second stage due to the subtle movements involved in the geometric reorganization has a very complex energy profile and therefore only a scan of the C−C bond distance is presented (see the Supporting Information for details of the complete exploration of the potential-energy surface). It implies a reorientation of all three benzene rings located in the catalyst, in the electrophile, and in the nucleophile and involves coordinated rotations of the different dihedral angles. The intramolecular attack of the nitronate intermediate generated on the ketimine is hindered because the orientation of the phenyl ring of the ketimine blocks this addition, thus preventing the formation of a pyrrolidine ring by a formal 1,3-dipolar cycloaddition. In this step, the coordination of the nucleophile 1 b to the catalyst 3 a is also crucial for the configuration of the products.19 Overall, the rate-limiting step is the proton transfer, which has a larger energy barrier than the C−C bond formation (see Figure 2).

Relative Gibbs free energy and energy profile of the two-step mechanism.
In summary, an organocatalytic strategy for the synthesis of α,γ-diamino acid derivatives with high ee values was developed. The key to the success is intramolecular activation through hydrogen bonding by an ortho hydroxy group, which allowed the Michael addition of glycine ylides bearing only one activating group.
Acknowledgements
The Spanish Government (CTQ2015-64561-R, CTQ2016-76061-P, MDM-2014-0377), the ERC (contract number: 647550), and CCC-UAM (computing time) are acknowledged. A.G.-C. and F.E. thank MINECO for PhD fellowships (FPI), and A.M.-S. thanks CAM for a postdoctoral contract (2016-T2/IND-1660).
Conflict of interest
The authors declare no conflict of interest.

