Totalsynthese von (+)-Rubriflordilacton A
Abstract
Zwei enantioselektive Totalsynthesen des Nortriterpenoid-Naturstoffs Rubriflordilacton A werden beschrieben. Sie verlaufen über Palladium- oder Cobalt-katalysierte Cyclisierungsrouten zu den CDE-Ringen und werden kurz vor Ende der Syntheseroute zusammengeführt. Das Grundgerüst für den Schlüsselschritt wird durch die konvergente Kupplung einer gemeinsamen Diinkomponente mit einem entsprechenden AB-Ring-Aldehyd gebildet. Eine Strategie, die den Weg für die Synthesen anderer Mitglieder der Naturstofffamilie ebnet.
Die chinesischen Kräuterpflanzen der Schisandra- und Kadsura-Gattung enthalten eine Vielfalt an strukturell ähnlichen Nortriterpenoid-Naturstoffen, charakterisiert durch ein komplexes fusioniertes Ringsystem, einen hohen Sauerstoffgehalt und eine Vielzahl stereogener Zentren.1 Viele dieser Verbindungen zeigen biologische Aktivität, unter anderem eine vielversprechende Wirkung gegen HIV. Ihre interessante Architektur stellt Synthesechemiker vor eine präparative Herausforderung, die zum ersten Mal 2011 von Yang und Mitarbeitern mit der Synthese von Schindilacton A angenommen wurde.2 Komplementiert wurde dieser Meilenstein kürzlich durch die elegante asymmetrische Synthese von Rubriflordilacton A (1; Schema 1) durch Li et al., bei der eine 6π-Elektrocyclisierung genutzt wurde, um den anspruchsvollen pentasubstituierten D-Arylring aufzubauen,3 sowie durch die Synthese der verwandten Schilancitrilactone B und C und Propindilacton G.4 Hier präsentieren wir zwei konvergente enantioselektive Totalsynthesen von Rubriflordilacton A,5 bei denen im Unterschied zu bisherigen Studien das CDE-Ringsystem in einer einzigen Tricyclisierungsstufe gebildet wird.6, 7 Die beiden Synthesen unterscheiden sich in der Methode, mithilfe derer das CDE-Gerüst aufgebaut wird: entweder durch Palladium- oder durch Cobalt-Katalyse. Die Produkte dieser Schlüsselschritte werden darauffolgend zu einem gemeinsamen Intermediat umgesetzt.

Retrosynthese.
Beide Strategien (Schema 1) streben in der letzten Stufe die Einführung des Butenolid-G-Rings via Addition eines Siloxyfurannukleophils 2 an ein Oxocarbeniumion an. Das letztgenannte Synthon würde sich aus Lactol 3 ergeben, das wiederum aus einem Bromendiin (4) oder Triin (5) unter Palladium- bzw. Cobaltkatalyse entsteht. Bromendiin 4 könnte durch die Kupplung von Aldehyd 66a mit Diin 7 gebildet werden, das zwei anspruchsvolle, aufeinander folgende Stereozentren aufweist. Die Synthese des Triins 5 würde ebenfalls 7 sowie einen Alkin enthaltenden Aldehyd 8 benötigen. Die Strategien weisen verschiedene Herausforderungen auf: Palladium-katalysierte Bromendiin-Cyclisierungen sind für die effiziente Synthese tricyclischer Systeme etabliert,8, 9 fanden bisher aber keine Anwendung in der Naturstoffsynthese. Die Cobalt-katalysierte Alkincyclotrimerisierung hat eine bedeutende Historie in der Synthesechemie,10 jedoch ist ihre Verwendung bei der Bildung von Siebenringen (wie der C-Ring in Rubriflordilacton A) selten.
Die Synthese von Diin 7 begann mit der Veresterung von (S,E)-Pent-3-en-2-ol11 mit Carbonsäure 9 (Schema 2).12 Eine Ireland-Claisen-Umlagerung des resultierenden Esters 10 lieferte Säure 11. Die Reaktion ergab eine höhere Ausbeute und Diasteroselektivität mit dem freien Lithiumenolat (96 %, d.r.>20:1)13 als mit dem Silylketenacetal (92 %, d.r. 9:1).12 Anschließende Umwandlungen der Carbonsäure in 11 in ein Benzyldimethylsilylalkin 12 (wobei sich die Stork-Zhao-Olefinierung14/Eliminierung als effizienteste Alkinierung erwies) und des para-Methyoxybenzylethers in ein terminales Alkin ergaben 7, vorbereitet für die Addition an die AB-Ring-Aldehyde 6 oder 8.

Reaktionsbedingungen: a) (S,E)-Pent-3-en-2-ol, EDC⋅HCl, Et3N, DMAP, THF, RT, 16 h, 81 %; b) LiHMDS, Et3N/Toluol (3:1), −78 °C→RT, 5 h, 95 %, d.r.>20:1; c) LDA, TMSCl/Et3N (1:1), THF, −78 °C→0 °C, 3 h, 92 %, d.r. 9:1; d) TMSCHN2, Toluol/MeOH (5:1), RT, 30 min, 88 %; e) DIBALH, CH2Cl2, −78→−30 °C, 2 h, 97 %; f) DMP, NaHCO3, CH2Cl2, 0 °C→RT, 1 h, 90 %; g) [Ph3PCH2I]+I−, NaHMDS, THF, −78 °C→RT; dann NaHMDS, −78 °C→RT, 84 %; h) LiHMDS, THF, −78 °C, 30 min; dann BnMe2SiCl, −78 °C→RT, 3 h, 98 %; i) DDQ, CH2Cl2/H2O (4:1), RT, 1 h; j) DMP, NaHCO3, CH2Cl2, 0 °C→RT 30 min, 83 % (über 2 Stufen); k) CBr4, PPh3, CH2Cl2 −30→0 °C, 1 h, 85 %; l) nBuLi, THF, −78 °C→RT, 40 min, 98 %. Bn=Benzyl, DIBALH=Diisobutylaluminiumhydrid, DDQ=2,3-Dichlor-5,6-dicyan-1,4-benzochinon, DMAP=4-Dimethylaminopyridin, DMP=Dess-Martin-Periodinan, EDC=1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid, HMDS=Hexamethyldisilazid, LDA=Lithiumdiisopropylamid, PMB=para-Methoxybenzyl, TMS=Trimethylsilyl.
Die Herstellung von Aldehyd 8 machte es möglich, eine Stille-Kupplung zu vermeiden, die bei der Synthese von Aldehyd 6 angewendet wird,6a da ein Alkin- und nicht eine Bromalkenseitenkette benötigt wird. Der erste Schritt erfolgte ausgehend von Ester 13 (Schema 3), der über Alkin-Carbocuprierung15 mit 3-Trimethylsilylpropinylmagnesiumbromid,16 Esterreduktion und asymmetrische Sharpless- Epoxidierung zum Epoxid 14 umgesetzt wurde (83 % über 3 Stufen, 92 % ee).17 Ringöffnung von 14 mit Allylmagnesiumchlorid,18 gefolgt von Oxidation und β-Lactonbildung, lieferte 15 in guten Ausbeuten (74 % über 4 Stufen).19 Durch zweifache nukleophile Addition von Methylmagnesiumbromid an das β-Lacton wurden die geminalen Dimethylgruppen des B-Rings (16) eingeführt.20 Oxidative Spaltung des terminalen Alkens in 16, Methylacetalbildung und Oxidation des verbliebenen primären Alkohols ergaben die Aldehydregioisomere 17 und 18 (Verhältnis 1.9:1, 72 % Ausbeute ausgehend von 16). Die Bildung der trennbaren Mischung der Aldehyde ist nicht von Bedeutung, da beide für die Umsetzung zum AB-Ring-Aldehyd 8 geeignet sind. Im Fall von Aldehyd 17 wurde durch Ando-Olefinierung/A-Ring-Lactonisierung21 und Acetalhydrolyse zunächst Lactol 19 hergestellt. Die Reaktion dieses Lactols mit Kaliumcarbonat in Methanol-Lösung lieferte den AB-Ring-Aldehyd 8 in quantitativer Ausbeute durch Oxy-Michael-Addition. Eine ähnliche Sequenz, die über das Enoat 20 (Z/E=2.5:1) verläuft, konnte ausgehend von Aldehyd 18 realisiert werden. Saure Entschützung der Acetale in 20 führte durch spontane Lactonisierung zu 19.

Reaktionsbedingungen: a) TMSCCCH2MgBr, CuBr⋅SMe2, THF, −78→−40 °C, 40 min; 13, −78 °C; b) DIBALH, CH2Cl2, −78 °C→RT, 3 h, 90 % (2 Stufen); c) Ti(OiPr)4, D-(−)-Diethyltartrat, tBuOOH, 4-Å-MS, CH2Cl2, −20 °C, 22 h, 92 %, 92 % ee; d) AllylMgBr, THF, 0 °C, 10 min, 97 %; e) SO3⋅Pyr, DMSO, iPr2EtN, CH2Cl2, 0 °C→RT, 2 h; f) NaOCl, NaH2PO4, 2-Methylbut-2-en, tBuOH/H2O (3:1), RT, 18 h, 92 % (2 Stufen); g) BOPCl, Pyr, MeCN, RT, 3 h, 83 %; h) MeMgBr, THF, −5 °C→RT, 1.5 h, 64 %+ 31 % Keton, Gesamtausbeute von 75 % nach Rückgewinnung; i) OsO4, NaIO4, 2,6-Lutidin, Dioxan/H2O (4.6:1), RT, 2 h, 88 %; j) (±)-Camphersulfonsäure, MeOH, RT, 18 h, 98 %; k) SO3⋅Pyr, DMSO, iPr2EtN, CH2Cl2, 0–10 °C, 1 h, 84 %; l) (PhO)2POCH2CO2Et, KHMDS, THF, 0 °C; m) TFA, CH2Cl2, 0 °C, 15 min, 47 % (von 17 und 18); n) K2CO3, MeOH, RT, 2 h, 99 %. BOPCl=Bis(2-oxo-3-oxazolidinyl)phosphonsäurechlorid, MS=Molekularsieb, Pyr=Pyridin, TBS=tert-Butyldimethylsilyl, TFA=Trifluoressigsäure.
Mit den so erhaltenen Diin- und Aldehyd-Schlüsselkomponenten wurde schließlich deren Verknüpfung und Cyclisierung zum ABCDE-Ringsystem von Rubriflordilacton A untersucht (Schema 4). Wir begannen mit der Palladium-katalysierten Route, die durch Addition von Diin 7 an Bromalkenylaldehyd 6 Alkohol 21 ergab (67 %). Vorherige Studien in unserer Gruppe6b hatten gezeigt, dass eine Schützung des Propargylalkohols nötig ist, um hohe Ausbeuten bei der darauffolgenden Cyclisierung zu gewährleisten; darum wurde der Alkohol silyliert. Das resultierende Bromendiin cyclisierte durch Reaktion mit [Pd(PPh3)4] (10 Mol-%) und Triethylamin in Acetonitril in exzellenter Ausbeute (91 %). Die Oxidation des Arylbenzyldimethylsilans zum entsprechenden Phenol verlief problemlos,22 und nach benzylischer Desoxygenierung wurde das vollständig funktionalisierte ABCDE-Gerüst 23 isoliert.

Reaktionsbedingungen: a) nBuLi, 7, −78 °C; dann Zugabe von 6, −78→−10 °C, 2 h, 67 %; b) TBSOTf, 2,6-Lutidin, CH2Cl2, 0 °C→RT, 4 h, 75 %; c) [Pd(PPh3)4] (10 Mol-%), Et3N, MeCN, 80 °C, 18 h, 91 %; d) TBAF, THF, RT, 30 min; dann H2O2, KHCO3, MeOH, RT, 12 h; e) Et3SiH, ZnCl2, CH2Cl2, RT, 3 h; dann TBAF, THF, RT, 20 min, 51 % (2 Stufen); f) nBuLi, 7, −78 °C; dann Zugabe von 8, −78→−10 °C, 4 h, 85 %; g) [CpCo(CO)2] (20 Mol-%), PPh3 (40 Mol-%), PhCl, MW (300 W), 150 °C, 25 min, 67 %; h) TBAF, THF, RT, 30 min; dann H2O2, KHCO3, MeOH, RT, 12 h, 84 %; i) Et3SiH, ZnCl2, CH2Cl2, RT, 3 h, 77 %; j) TBSCl, Imidazol, DMAP, CH2Cl2, RT, 6 h, 98 %; k) [CpCo(CO)2] (20 Mol-%), PPh3 (40 Mol-%), PhCl, MW (300 W), 150 °C, 25 min, 54 %. Cp=Cyclopentadienyl, MW=Mikrowellen, OTf=Triflat, TBAF=Tetrabutylammoniumfluorid.
An dieser Stelle entschieden wir uns, die Palladium-katalysierte Cyclisierungsroute mit dem alternativen Cobalt-katalysierten Cyclotrimerisierungsansatz zu vergleichen. Das Diin 7 wurde daher mit Alkinylaldehyd 8 umgesetzt, wodurch Triin 24 gebildet wurde (85 %). Cyclotrimerisierung von 24 in der Mikrowelle6b, 23 lieferte den Pentacyclus 25 (67 %). Dieses Produkt kann zu derselben ABCDE-Ringstruktur 23 umgesetzt werden, die auch durch die Palladium-katalysierte Route gebildet wurde (durch Tamao-Oxidation, gefolgt von benzylischer Desoxygenierung).22 Zu erwähnen sei hier auch, dass die Silylierung des Propargylalkohols in 24 einen alternativen Schnittpunkt zwischen den beiden Routen darstellt, da das Produkt der Cyclotrimerisierung des resultierenden Silylethers der Pentacyclus 22 ist, wenngleich mit etwas geringer Effizienz gegenüber der Cyclisierung des freien Alkohols 24.24
Nachdem nun beide Strategien das gleiche Intermediat 23 aufwiesen, blieb noch der Aufbau des FG-Ringsystems. Dieser wurde in vier Stufen erreicht (Schema 5), beginnend mit einer zweistufigen oxidativen Spaltung der Alkenseitenkette in 23, die das Lactol 26 (im Gleichgewicht mit dem offenkettigen Aldehyd) lieferte. Dieses Lactol bildet einen Schnittpunkt mit der von Lis Gruppe berichteten Route,3 wo 26 durch die Bildung eines Fluorpyrans und anschließende stereoselektive Kupplung mit Furanylstannan zum Butenolid-G-Ring umgesetzt worden war. Um zinnbasierte Nukleophile zu vermeiden, untersuchten wir eine alternative Aktivierung des Lactols. Dies erwies sich als schwierig, aber nach einigen Experimenten stellten wir fest, dass Chlorpyran 27 durch Reaktion des Lactols 26 mit einer Mischung aus Thionylchlorid und Zink(II)-chlorid hergestellt werden kann.25 Die Umsetzung verläuft über das sich anfänglich schnell bildende Dimer 28, das binnen 3 h zum Chlorpyran 27 reagiert (siehe Diagramm in Schema 5). Dieses instabile Intermediat wurde direkt mit Siloxyfuran 28 in der Gegenwart von Zink(II)-chlorid umgesetzt und lieferte Rubriflordilacton A zusammen mit dem C23-Epimer 30 in 71 % Ausbeute (d.r.≈1:1). Bemerkenswert ist die exzellente faciale Selektivität der Addition des Furans an das Oxocarbeniumion, aufgrund derer die beiden trennbaren Diastereomere die hauptsächlichen Produkte der Addition sind. Die spektroskopischen Daten des synthetischen Rubriflordilactons A stimmen mit denen des Naturstoffes überein, mit der Ausnahme des spezifischen Drehwinkels, der einen ähnlichen Wert mit entgegengesetztem Vorzeichen aufweist. Dies lässt darauf schließen, dass 1 das nicht natürliche Enantiomer ist (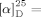 +58.3 (c=0.114, MeOH); Lit.:
+58.3 (c=0.114, MeOH); Lit.: 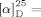 −58.1 (c=0.114, MeOH)).5, 26
−58.1 (c=0.114, MeOH)).5, 26

Reaktionsbedingungen: a) OsO4 (2 Mol-%), NMO, Aceton/H2O (3:1), RT, 3 h; b) NaIO4/SiO2, CH2Cl2, RT, 15 min, 85 % (2 Stufen); c) ZnCl2, SOCl2, CDCl3, RT, 3 h; d) 29, ZnCl2, CH2Cl2, −30 °C→RT, 12 h, 38 % von 1 und 33 % von 30 (2 Stufen). NMO=N-Methylmorpholin-N-oxid, TIPS=Triisopropylsilyl.
Zusammenfassend haben wir zwei Synthesestrategien zur enantioselektiven Herstellung von Rubriflordilacton A realisiert, die eine Palladium- oder eine Cobalt-Katalyse zum Aufbau des ABCDE-Ringsystems als Schlüsselschritt enthalten. Die Routen sind strategisch hochgradig konvergent, da sie erst vier Stufen vor Abschluss der Syntheseroute zusammenlaufen. Die Kupplung eines funktionalisierten Diins mit dem AB-Ring-Aldehyd zur Bildung des Cyclisierungssubstrats eröffnet die einzigartige Möglichkeit zur Synthese von weiteren Mitgliedern dieser faszinierenden Naturstofffamilie ebenso wie von Rubriflordilacton-Analoga.
Acknowledgements
Wir danken EPSRC für ein Forschungsstipendium (EAA) (EP/E055273/1) und zusätzliche Finanzierung (EP/K005391/1), A*STAR für ein National Science Stipendium (SSG) und dem DAAD (Postdoc-Stipendum für BG).