

Genetic variation at 16q24.2 is associated with small vessel stroke
Abstract
Objective
Genome-wide association studies (GWAS) have been successful at identifying associations with stroke and stroke subtypes, but have not yet identified any associations solely with small vessel stroke (SVS). SVS comprises one quarter of all ischemic stroke and is a major manifestation of cerebral small vessel disease, the primary cause of vascular cognitive impairment. Studies across neurological traits have shown that younger-onset cases have an increased genetic burden. We leveraged this increased genetic burden by performing an age-at-onset informed GWAS meta-analysis, including a large younger-onset SVS population, to identify novel associations with stroke.
Methods
We used a three-stage age-at-onset informed GWAS to identify novel genetic variants associated with stroke. On identifying a novel locus associated with SVS, we assessed its influence on other small vessel disease phenotypes, as well as on messenger RNA (mRNA) expression of nearby genes, and on DNA methylation of nearby CpG sites in whole blood and in the fetal brain.
Results
We identified an association with SVS in 4,203 cases and 50,728 controls on chromosome 16q24.2 (odds ratio [OR; 95% confidence interval {CI}] = 1.16 [1.10–1.22]; p = 3.2 × 10−9). The lead single-nucleotide polymorphism (rs12445022) was also associated with cerebral white matter hyperintensities (OR [95% CI] = 1.10 [1.05–1.16]; p = 5.3 × 10−5; N = 3,670), but not intracerebral hemorrhage (OR [95% CI] = 0.97 [0.84–1.12]; p = 0.71; 1,545 cases, 1,481 controls). rs12445022 is associated with mRNA expression of ZCCHC14 in arterial tissues (p = 9.4 × 10−7) and DNA methylation at probe cg16596957 in whole blood (p = 5.3 × 10−6).
Interpretation
16q24.2 is associated with SVS. Associations of the locus with expression of ZCCHC14 and DNA methylation suggest the locus acts through changes to regulatory elements. Ann Neurol 2017;81:383–394
Genome-wide association studies (GWAS) enable identification of common genetic variants that influence disease risk and have proved successful in elucidating pathophysiological mechanisms underlying diseases with a genetic influence.1 A number of GWAS associations have recently been identified with ischemic stroke, almost all of which have been associated with specific stroke subtypes.2-4 A number of genetic associations have been reported with cardioembolic (CE) and large artery stroke (LAS), but, in contrast, there have been no robust associations solely with small vessel stroke (SVS). This is despite epidemiological data that suggest genetic factors are particularity important for SVS. For example, there are a number of monogenic stroke disorders associated with SVS,5 and family history studies have shown a strong association between SVS and a family history of stroke.6 Similarly, related traits, including white matter hyperintensities (WMH), have been shown to have high heritability.7
SVS itself comprises one quarter of all ischaemic stroke and is one of the clinically overt manifestations of cerebral small vessel disease (SVD), the major cause of vascular cognitive impairment. Other radiological features of SVD include WMH, best observed on T2-weighted magnetic resonance imaging (MRI), cerebral microbleeds—observed on gradient echo MRI, and intracerebral hemorrhages (ICH).8 Despite its importance, the pathogenesis of SVD remains poorly understood and this limits the development of proven treatments for established disease.
One consistent finding across adult-onset neurological complex diseases, including Parkinson's disease,9 Alzheimer's disease,10 and stroke,11 is that younger onset cases have a stronger genetic burden from common disease-associated single-nucleotide polymorphisms (SNPs). Leveraging this increased burden, by focussing on younger-onset cases in analysis of genetic data, can lead to detection of novel trait-associated variants.11 This may be particularly relevant for SVS, given that epidemiological studies have shown stronger associations with SVS and a family history of stroke in younger stroke cases.6
Here, we perform an age-at-onset informed GWAS meta-analysis in stroke, including a large population of younger-onset (age < 70) SVS cases. We perform analysis for all ischemic stroke (IS) and its three subtypes: CE, LAS, and SVS. Using this approach, we identify a novel association with SVS, seek further validation of the locus in other SVD phenotypes, and assess the influence of SNPs at the locus on messenger RNA (mRNA) expression of nearby genes and DNA methylation at nearby CpG sites.
Materials and Methods
Study Design
We used a three-stage design for the association analysis (Fig 1). In brief, in stage I, we performed association analysis of stroke phenotypes in 10,210 cases and 12,285 controls of European ancestry from Europe, United States, and Australia; most of which contributed to the METASTROKE ischaemic stoke GWAS meta-analysis—and all of which have been described previously (Table 1).2, 12, 13 In all cases, diagnosis of stroke was based on clinical evaluation with radiological confirmation. Subtyping of stroke cases was based on the TOAST criteria; in this analysis, we considered the CE, LAS, and SVS subtypes.14 Of note, our SVS analysis included a large sample (1,012 cases, 970 controls) of younger-onset (age < 70) MRI-confirmed lacunar strokes, meaning that, although we investigated all subtypes, we had most power to identify associations with SVS.

Flow chart of analyses performed. GTEx = Genotype-Tissue Expression; mRNA = messenger RNA; SNP = single-nucleotide polymorphism; SVS = small vessel stroke; [Color figure can be viewed at wileyonlinelibrary.com]
| Population | IS | CE | LAS | SVS | Controls | % Cases With MRI | Age of Cases (mean (SD)) |
|---|---|---|---|---|---|---|---|
| Stage I populations | |||||||
| ASGC | 1,162 | 240 | 421 | 310 | 1,244 | 43.0 | 72.9 (13.2) |
| WTCCC2-Germany | 1,174 | 330 | 346 | 106 | 797 | 83.0 | 66.7 (12.9) |
| WTCCC2-UK | 2,374 | 474 | 498 | 460 | 5,175 | 37.2 | 72.2 (12.5) |
| Milano | 366 | 64 | 73 | 25 | 407 | 86.7 | 57.4 (15.6) |
| DNA-lacunar/GENESIS | 1,287 | 80 | 64 | 1,012 | 970 | 100.0 | 59.6 (12.0) |
| LSS | 455 | 157 | 70 | 55 | 455 | 89.0 | 67.7 (14.5) |
| ISGS/SWISS | 1,014 | 235 | 217 | 187 | 1,370 | 83.0 | 66.5 (13.6) |
| BRAINS | 361 | 29 | 120 | 97 | 444 | 30.8 | 74.4 (14.2) |
| MGH-GASROS | 294 | 106 | 68 | 23 | 376 | 60.0 | 66.7 (14.5) |
| VISP | 1,723 | — | — | — | 1,047 | 47.0 | 68.0 (10.7) |
| Total (discovery) | 10,210 | 1,715 | 1,877 | 2,275 | 12,285 | ||
| Stage II populations | |||||||
| NINDS Stroke Genetics Network | 7,743 | 2,001 | 1,130 | 1,408 | 17,970 | 62.0 | 66.3 (14.8) |
| Stage III populations | |||||||
| deCODE | — | 1,100 | — | 520 | 20,473 | NA | 72.7 (11.6) |
| Total | 17,953 | 4,816 | 3,007 | 4,203 | 50,728 | ||
- IS = all ischemic stroke; CE = cardioembolic stroke; LAS = large artery stroke; SVS = small vessel stroke; ASGC = Australian Stroke Genetics Collaborative; WTCCC2 = Wellcome Trust Case Control Consortium 2; LSS = Leuven Stroke Study; BRAINS = Bio-repository of DNA in stroke; MGH-GASROS = The MGH Genes Affecting Stroke Risk and Outcome Study; VISP = The Vitamin Intervention for Stroke Prevention Trial; NA = information not available.
In stage II, we took three SNPs from the top 25 loci from each phenotype forward for a first in silico replication in the NINDS Stroke Genetics Network (SiGN),2 which consisted of 7,743 cases and 17,790 controls. We meta-analyzed stages I and II together and identified three loci with p < 5 × 10−7. Finally, in stage III, we determined whether these three SNPs were associated with the phenotype in which they were identified (CE or SVS) by in silico replication in a large Icelandic population (deCODE; 520 SVS cases, 1,100 CE cases, 50,728 controls; stage III).
Genotyping and Imputation
Genotyping, quality control, and imputation of all studies has been described previously.2, 3, 13 All studies were genotyped on commercially available arrays from Illumina (San Diego, CA) or Affymetrix (Santa Clara, CA) and imputed to 1000 Genomes phase 1 reference panels using IMPUTE or MACH.15 Imputation quality score was assessed by calculating the ratio of the observed to the expected binomial variance of the allele dosage.
Association Analysis
Association analysis was performed using a covariate-informed approach,11, 16 which we, and others, have implemented previously.11, 17 Briefly, the approach uses case/control status and a covariable—in this case, age-at-onset—to estimate each individual's stroke liability, which can be interpreted as their underlying propensity to stroke, on a normally distributed scale. In this analysis, cases with an earlier age-at-onset take more-extreme positive values than late-onset cases given that, attributed to the lower prevalence of stroke at younger ages, they are assumed to have higher stroke liability. Conversely, controls who are older and stroke free at age-at-observation take more-extreme negative value than younger controls given that they have been stroke free for a longer time and are therefore assumed to have a lower stroke liability.
In this analysis, the approach was implemented in our software, CIAO (provided at https://sites.google.com/site/mtraylor263/software/covariate-informed-gwas-analysis). Specifically, the approach taken is to model phenotype data using a continuous unobserved normally distributed quantitative trait, called the disease liability (
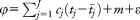 ), where
), where
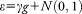 and
and
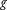 denotes the genetic effects. Then, an individual is a case (z = 1) if and only if
denotes the genetic effects. Then, an individual is a case (z = 1) if and only if
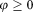 and is a control (z = 0) otherwise.
and is a control (z = 0) otherwise.
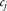 is a parameter estimating the effect of a given covariate j on the liability scale.
is a parameter estimating the effect of a given covariate j on the liability scale.
 denotes the disease prevalence p at the covariate mean
denotes the disease prevalence p at the covariate mean
 under a normal cumulative distribution function
under a normal cumulative distribution function
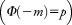 . This model is used to approximate the effect of a disease covariate—in this case, age-at-onset—on the liability scale, based on estimates of risk of IS by age from epidemiological data, thereby estimating
. This model is used to approximate the effect of a disease covariate—in this case, age-at-onset—on the liability scale, based on estimates of risk of IS by age from epidemiological data, thereby estimating
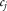 . For this analysis, the sex-specific risk of IS by age from an index age of 55 was obtained from population-based estimates (1.8%, 5.4%, and 12.1% before 65, 75, and 85, respectively, in women; 2.4%, 7.3%, and 12.6% before 65, 75, and 85, respectively, in men).18 We assumed that 20% of IS cases had each of the cardioembolic, small vessel, or large vessel stroke subtypes, approximating proportions observed in population-based studies.19 We developed two models for our analysis; one based on the risk rates for all IS and, second, for the three stroke subtypes. We used these models to calculate posterior mean liabilities after conditioning on age-at-onset for the four stroke phenotypes separately (
. For this analysis, the sex-specific risk of IS by age from an index age of 55 was obtained from population-based estimates (1.8%, 5.4%, and 12.1% before 65, 75, and 85, respectively, in women; 2.4%, 7.3%, and 12.6% before 65, 75, and 85, respectively, in men).18 We assumed that 20% of IS cases had each of the cardioembolic, small vessel, or large vessel stroke subtypes, approximating proportions observed in population-based studies.19 We developed two models for our analysis; one based on the risk rates for all IS and, second, for the three stroke subtypes. We used these models to calculate posterior mean liabilities after conditioning on age-at-onset for the four stroke phenotypes separately (
 ). Controls were modeled in the same way, but were assumed to take the posterior mean from the lower (unaffected) portion of the distribution in the liability threshold model (
). Controls were modeled in the same way, but were assumed to take the posterior mean from the lower (unaffected) portion of the distribution in the liability threshold model (
 . Where age data was missing, individuals were assigned the median age value (<1% of cases). Regression was then performed on posterior liabilities (
. Where age data was missing, individuals were assigned the median age value (<1% of cases). Regression was then performed on posterior liabilities (
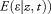 by multiplying the number of samples by the squared correlation between the expected genotype dosage and posterior mean liabilities for each of the discovery cohorts in the four IS phenotypes (all IS, CE, LAS, and SVS). Ancestry-informative principal components were included, where appropriate, using the EIGENSTRAT procedure.20 Any residual inflation was accounted for by adjusting results by the genomic inflation factor, λ.21 In all analyses, SNPs with imputation quality score <0.7 or minor allele frequency (MAF) < 0.01 were excluded and meta-analysis was performed using Stouffer's method in METAL.22
by multiplying the number of samples by the squared correlation between the expected genotype dosage and posterior mean liabilities for each of the discovery cohorts in the four IS phenotypes (all IS, CE, LAS, and SVS). Ancestry-informative principal components were included, where appropriate, using the EIGENSTRAT procedure.20 Any residual inflation was accounted for by adjusting results by the genomic inflation factor, λ.21 In all analyses, SNPs with imputation quality score <0.7 or minor allele frequency (MAF) < 0.01 were excluded and meta-analysis was performed using Stouffer's method in METAL.22
Further Analysis of a Novel Locus Associated With SVS
For a novel variant associated with SVS, we performed further analysis to elucidate the association for different groups based on age-at-onset. First, for data sets in stage I and II, we divided the cases into quartiles based on age-at-onset and estimated the association of the SNP with each quartile using logistic regression with all controls, meta-analyzing using a fixed-effects inverse variance weighted approach (data not available in BRAINS, MGH-GASROS, and ISGS/SWISS). Second, we interrogated associations at the locus in non-European ancestry populations, comprising 657 small vessel African-American stroke cases and 3,251 matched controls from the NINDS Stroke Genetics Network and African or African-Caribbean ancestry individuals from the South London Ethnicity and Stroke Study (SLESS),2, 23 and 314 SVS cases and 5,193 controls of Pakistani ancestry from the RACE study.3 We used logistic regression to evaluate the association within each group and evaluated the overall transethnic association by meta-analyzing using Stouffer's method.
In addition, we explored the association of the SNP with other SVD phenotypes. We evaluated association of the SNP with (1) white matter hyperintensity volumes (WMHV) measured on T2-weighted MRI in 3,670 IS patients of European ancestry24; (2) in MRI-defined small subcortical brain infarcts (SSBI) brain infarcts in 17,197 transethnic individuals (85.7% European; 8.8% African-American; 3.5% Hispanic; 1.0% Chinese; and 1.0% Malay) from community studies recruited within the neuro-CHARGE consortium (mean age, 68.90 ± 10.31; 1,986 with infarcts). SSBI were defined as MRI-defined brain infarcts of 3 to 15 or 3 to 20mm in size, located in the basal ganglia, the white matter, or the brainstem. Association analysis was performed overall, and for the subset of cases with extensive WMH burden—defined as the top age-specific quartile of WMHV on a quantitative scale or above the age-specific median by 5-year age categories for studies using semiquantitative measurements of WMH burden; N = 549; and (3) ICH in 1,545 European ancestry cases and 1,481 controls, described previously,25 and stratified according to lobar or nonlobar location.
Evaluation of Regulatory Chromatin States, mRNA Expression, and DNA Methylation
To investigate a novel locus, we used existing resources and performed some further analyses to characterize its regulatory potential. We interrogated chromatin states and regulatory motifs from ENCODE and Epigenomics Roadmap using Haploreg v4.1.26 We also evaluated whether the associated SNP influences gene expression using the Genotype-Tissue Expression (GTEx) portal.27 Upon identifying an association between the SNP and expression of a nearby gene, we evaluated the evidence that the association signal for SVS and gene expression derives from the same causal variant using a Bayesian colocalization test.28 Using the R coloc package (http://cran.r-project.org/web/packages/coloc), we compared five models for SNPs with 50kb of our lead SNP using the approach (H0: No association with either trait; H1: Association with SVS, not with expression; H2: Association with expression, not with SVS; H3: Association with SVS and expression, two independent SNPs; H4: Association with SVS and expression, one shared SNP).
Next, we assessed whether the lead SNP (rs12445022), or three SNPs in linkage disequilibrium (LD; rs4843625, rs12920915, and rs12444224), influence DNA methylation levels in whole blood. We evaluated genetic associations of whole-blood DNA methylation levels at selected CpG sites profiled on the Illumina Infinium HumanMethylation450 BeadChip array in a group of 660 monozygotic (MZ) female twins (mean age, 59; age range, 18–79). These individuals were research volunteers from the TwinsUK cohort in the United Kingdom.29 All were of European ancestry. For each CpG site of interest, we calculated the normalized methylation means for the 330 MZ twin pairs as a phenotype in the genetic analysis and took into account covariates, including smoking, body mas index, age, methylation plate, and blood-cell count estimates. TwinsUK imputed genotypes were obtained for the 1000 Genomes reference set,30 where we excluded SNPs with Hardy–Weinberg p < 1 × 10−4, MAF < 5%, and those with IMPUTE info value <0.8. We tested for association with our SNP, or SNPs in close LD (r2 > 0.6) with DNA methylation at CpG sites. We used p < 4 × 10−5, equivalent to a false discovery rate (FDR) < 5%,31 to identify significant cis-mQTL associations.
Finally, we explored genetic associations at 16q24.2 (defined as within 50kb of rs12445022) with DNA methylation profiles in 166 human fetal brain samples (92 male, 74 female) ranging from 56 to 166 days postconception initially using publically available data—which hold results for mQTL associations reaching the study-wide significance threshold (http://epigenetics.essex.ac.uk/mQTL/). Methods for this study have been published in detail elsewhere.32 Briefly, DNA methylation levels were profiled on the Illumina Infinium HumanMethylation450 BeadChip array and SNP genotypes were obtained from the Illumina HumanOmniExpress BeadChip and imputed to 1000 Genomes phase 3 using SHAPEIT and Minimac3 through the Michigan Imputation Server.15, 33 SNP-methylation probe pairs were tested using the R package, MatrixEQTL,34 including covariates to control for age, sex, and ancestry-informative principal components. Upon identifying a significant association at 16q24.2, we performed additional analyses (not publicly available: we gained access to the data) to test whether any of our four SNPs (rs12445022, rs4843625, rs12920915, and rs12444224) were associated with methylation at the identified probe. We again used p < 4 × 10−5, equivalent to an FDR <5%,31 to identify significant cis-mQTL associations.
Results
Association Analysis
In phase I association analysis, we confirmed previous associations between HDAC9 and LAS (rs2107595, p = 3.0 × 10−8) and between PITX2 and CE (rs192172299, p = 2.0 × 10−9).3, 4 Previous associations between ZFHX3 and CE and between MMP12 and LAS did not reach genome-wide significance in this analysis (rs879324, p = 5.0 × 10−7 and rs586701, p = 0.0014, respectively).11 An SNP in a region close to HABP2 previously associated with young-onset IS was also significant, albeit not genome wide, in this analysis (rs11196288; p = 2.4 × 10−4).35 Genomic inflation λ and the equivalent values, scaled to 1,000 cases and 1,000 controls (λ1000),36 were well controlled across all analyses (IS, λ (λ1000) = 1.05 (1.00); CE, λ (λ1000) = 1.02 (1.00); LAS, λ (λ1000) = 1.02 (1.00); SVS. λ (λ1000) = 1.01 (1.00)).
We took 25 independent loci forward (three SNPs in LD from each locus selected on p value) from each analysis (IS, CE, LAS, and SVS) for in silico replication in the NINDS Stroke Genetics Network study (stage II). Information on these SNPs is provided in Supplementary Tables 1 to 4. Following this analysis, excluding previously reported associations, three loci showed significance at p < 5 × 10−7 (two with SVS, one with CE) and one was genome-wide significant (rs12445022, p = 4.4 × 10−8, associated with SVS). We followed up all three loci in a second in silico replication (stage III) in a large Icelandic population (deCODE). A single SNP, rs12445022, showed evidence of replication (p = 0.011). When performing a meta-analysis across all populations, rs12445022 was associated with SVS at genome-wide significance (p = 3.2 × 10−9; Fig 2). The SNP was either genotyped or well imputed (info > 0.9) in all cohorts and lies in an intergenic region between junctophilin 3 (JPH3) and zinc finger CCHC domain-containing 14 (ZCCHC14). To confirm the association with rs12445022, we repeated the analysis using logistic regression, the approach taken in a conventional GWAS. The association was validated using this method, and associations were consistent across populations (odds ratio [OR; 95% confidence interval {CI}] = 1.16 [1.10–1.22]; p = 1.3 × 10−8; heterogeneity, p = 0.56; Fig 3).

Associations at 16q24.2 with (A) small vessel stroke, (B) cerebral white matter hyperintensities, (C) mRNA expression of ZCCHC14, and (D) gene locations and associations of the locus with DNA methylation. mRNA = messenger RNA; SVS = small vessel stroke; WMH = white matter hyperintensities; ZCCHC14 = zinc finger CCHC domain-containing 14; JPH3 = junctophilin 3; meQTL = methylation quantitative trait locus. [Color figure can be viewed at wileyonlinelibrary.com]

Forest plot of associations with rs12445022 under a logistic regression model. CI = confidence interval. [Color figure can be viewed at wileyonlinelibrary.com]
Further Analysis of a 16q24.2 Novel Locus Associated With SVS
We evaluated association of the lead SNP in different quantiles of age at stroke onset, using all controls in each analysis. The strongest associations were observed in younger-onset cases, suggesting that the influence of the SNP might be greatest in these individuals (Fig 4). However, this was not demonstrated statistically (p > 0.05).

Association of rs12445022 with small vessel stroke by quartiles of age-at-stroke onset in Europeans. CI = confidence interval.
We performed further analysis to assess whether the SVS-associated SNP influenced other manifestations of cerebral SVD. The SNP (rs12445022) was also associated with increased T2-WMHV (OR [95% CI] = 1.10 [1.05–1.16]; p = 5.3 × 10−5; Figs 2 and 5) and showed little heterogeneity across study groups (heterogeneity, p = 0.58). Conversely, the SNP was not associated with ICH—neither overall, nor in subgroups divided by lobar/nonlobar location. For SSBI, the direction of effect was the same as for SVS, but the effect was weaker and nonsignificant (OR [95% CI] = 1.05 [0.97–1.14]; p = 0.28). For the subgroup with WMH, the effect was stronger—and similar to that observed for SVS, but was again nonsignificant (OR [95% CI] = 1.15 [0.99–1.33]; p = 0.076).

Associations with rs12445022 for stroke and cerebral small vessel disease phenotypes. CI = confidence interval; IS = all ischemic stroke; WMH = white matter hyperintensities.
We next evaluated the identified locus in non-European ancestry populations. The SNP had a similar frequency to Europeans in South Asians from RACE (MAF = 37%), but was rarer in African ancestry populations, consisting of African Americans from the NINDS Stroke Genetics Network and United Kingdom individuals of African or African-Caribbean ethnicity from SLESS (MAF = 14%). Associations with the SNP were in the same direction as in European ancestry populations (Fig 4), but did not reach statistical significance in either ancestry, reflecting the much smaller sample sizes. However, when combining data from all populations, evidence for association at the SNP (p = 1.4 × 10−9) was stronger than in European ancestry populations alone, which might suggest a common association across populations. Indeed, there was no evidence of a significant difference in the strength of association between the European and non-European ancestry individuals (p = 0.64).
Regulatory Chromatin States, mRNA Expression, and DNA Methylation Related to 16q24.2
We used existing databases to assess the functional consequences of SNPs in the 16q24.2 region. First, we used the Haploreg v4.1 database to interrogate chromatin states and regulatory motifs from ENCODE and the NIH Roadmap Epigenomics Mapping Consortium.26, 37, 38 The database showed that our lead SNP influences chromatin states in multiple tissues. The SNP is classified as a genic promoter in nine tissues, an enhancer in 13 tissues, and overlaps DNAse1 hypersensitivity sites in 21 tissues.
Second, we used publicly available databases to evaluate the evidence that the lead SNP influences expression of nearby genes using the GTEx portal.27 The implicated A allele of our lead SNP (rs12445022) was associated with decreased expression of ZCCHC14 in tibial arterial tissue (p = 9.4 × 10−7; Fig 2). We used a Bayesian colocalization technique to assess whether the same variant drives the both the SVS association signal and mRNA expression of ZCCHC14.28 There was overwhelming evidence in support of H4 (posterior probability = 99.7%), strongly indicating that a single variant—most likely to be rs12445022—influences both SVS and expression of ZCCHC14.
Finally, we performed analyses to assess whether the lead SNP, or the three SNPs in LD, influence DNA methylation at CpG probes in whole blood. We found evidence that the lead SNP, and three SNPs in close LD (r2 > 0.6), influence DNA methylation at 4 nearby CpG sites (cg16596957, cg10312981, cg03020503, and cg00555085; all p < 4.0 × 10−5; Table 2). The implicated A allele of rs12445022 was associated with decreased methylation at the cg16596957 probe (beta [standard error; SE] = –0.38 [0.082]; p = 5.3 × 10−6). The SNPs explained between 5% and 8% of the methylation variance at the given CpG sites. The same 16q24.2 region by CpG probe (cg16596957) association was also recently reported in another study in whole blood.40 In addition, we looked for an association between SNPs at the 16q24.2 locus and DNA methylation levels in fetal brains, initially using publicly available data (http://epigenetics.essex.ac.uk/mQTL/). There was a strong association with SNPs in distant LD with our lead SNP (rs8047314 ∼ cg08031982; p = 7.1 × 10−14; r2 = 0.16 with rs12445022). We then performed additional analyses (not publicly available: we gained access to the data) to test whether our lead SNP, or the SNPs in close LD, were associated with methylation at cg08031982. We could identify no associations that reached our significance threshold (p < 4.0 × 10−5). However, there was a near-significant association of rs12920915 and rs4843625 with methylation at the cg08031982 probe (both p = 7.8 × 10−5). Our lead SNP, rs12445022, was not associated (p = 9.9 × 10−4).
| SNP Variant | SNP BP | CpG Probe | Probe BP | RA | Beta (SE) | r2 | p |
|---|---|---|---|---|---|---|---|
| rs12445022 | 87,575,332 | cg16596957 | 87,575,151 | A | –0.38 (0.082) | 0.058 | 5.3 × 10−6 |
| rs4843625 | 87,576,996 | cg16596957 | 87,575,151 | C | –0.33 (0.075) | 0.053 | 1.3 × 10−5 |
| rs4843625 | 87,576,996 | cg10312981 | 87,577,304 | C | 0.39 (0.074) | 0.077 | 1.9 × 10−7 |
| rs4843625 | 87,576,996 | cg03020503 | 87,577,656 | C | 0.35 (0.075) | 0.059 | 5.0 × 10−6 |
| rs4843625 | 87,576,996 | cg00555085 | 87,616,248 | C | 0.34 (0.075) | 0.057 | 6.6 × 10−6 |
| rs12920915 | 87,577,521 | cg16596957 | 87,575,151 | T | -0.38 (0.075) | 0.069 | 7.3 × 10−7 |
| rs12920915 | 87,577,521 | cg10312981 | 87,577,304 | T | 0.38 (0.075) | 0.068 | 1.0 × 10−6 |
| rs12920915 | 87,577,521 | cg03020503 | 87,577,656 | T | 0.34 (0.076) | 0.055 | 1.1 × 10−5 |
| rs12920915 | 87,577,521 | cg00555085 | 87,616,248 | T | 0.33 (0.076) | 0.051 | 2.2 × 10−5 |
| rs12444224 | 87,580,855 | cg16596957 | 87,575,151 | T | –0.38 (0.075) | 0.068 | 8.0 × 10−7 |
| rs12444224 | 87,580,855 | cg10312981 | 87,577,304 | T | 0.38 (0.075) | 0.069 | 8.7 × 10−7 |
| rs12444224 | 87,580,855 | cg03020503 | 87,577,656 | T | 0.35 (0.076) | 0.054 | 1.1 × 10−5 |
| rs12444224 | 87,580,855 | cg00555085 | 87,616,248 | T | 0.32 (0.076) | 0.050 | 2.6 × 10−5 |
- SNP = single-nucleotide polymorphism; BP = base position; RA = reference allele; SE = standard error; r2 = proportion of methylation variance explained by respective genotype.
Discussion
Genome-wide association studies in SVS have largely been disappointing. Some studies have suggested that an association with all IS at the highly pleiotropic 12q24.12 is driven by an association with SVS,2 but no genome-wide significant associations specifically with SVS have yet been identified. Using an age-of–onset informed analysis approach, we identified a novel locus at 16q24.2 associated with SVS. The SNP was also associated using a standard logistic regression approach, but was less significant—a difference of almost an order of magnitude (p = 3.2 × 10−9 compared to p = 1.3 × 10−8). In addition, the association was stronger with younger-onset SVS, suggesting a greater influence in these individuals. We tested whether the 16q24.2 association extends to other cerebral SVD-related phenotypes. We showed that the same locus also influences WMH and may have a similar effect on MRI-defined subcortical brain infarcts from prospective studies, although the association did not reach significance in our analysis. However, the locus does not appear to influence risk of ICH. A SNP in the same 16q24.2 region (rs4081947), in partial LD with our SNP (r2 = 0.28), was also recently reported to be associated with migraine in a large GWAS meta-analysis.41 These data provide strong supportive evidence that this 16q24.2 locus harbors variants that influence diseases of the cerebral vasculature.
Identifying the mechanisms by which GWAS associations influence disease risk presents additional challenges. In this case, the underlying mechanism and the specific genes implicated remains uncertain. Interrogation of mRNA expression data points to the lead SNP influencing expression of the nearest gene, ZCCHC14. This gene is ubiquitously expressed, but is highly expressed in arterial tissues and in the brain. However, its function is not well characterized. Zinc fingers of the CCHC-type contain an 18-digit residue found in the nucleocapsid of retroviruses and therefore may be important in viral response. Other plausible candidate genes reside nearby. The locus lies around 1Mb away from genes encoding forkhead box proteins, including FOXC2, FOXL1, and FOXF1. These proteins, particularly the closely related FOXC1—a paralogue of FOXC2, have been implicated in Mendelian forms of SVS.42 We found no evidence linking our SNP to expression of these genes. However, the function of these proteins changes dramatically between early development and in adult tissues,43 which might explain the absence of an association. This, coupled with the fact that FOXF2 variants have also recently been implicated in IS,44 make forkhead box proteins exciting targets for follow-up experiments.
Assessing DNA methylation, the process by which methyl groups are added to DNA thereby modifying its function, offers another potential method for mechanisticinsight. This epigenetic process influences gene expression and regulation in humans, and may be particularly relevant for diseases, such as stroke, where gene-environment interactions are likely to play an important role in pathogenesis.45 Substantial interindividual variation exists with respect to age and tissue type.46 However, an important emerging mechanism influencing methylation is local sequence content.47 Notably, recent studies have shown that GWAS findings from stroke-relevant traits, such as blood pressure, are likely to act by influencing DNA methylation.48 This may be particularly relevant for SVS, in which environmental and other vascular risk factors, such as hypertension, are important and have been shown to interact with disease risk.49 We evaluated whether our associated SNP (rs12445022), or SNPs in close LD, influence methylation of nearby CpG sites. We found evidence from whole blood that the same genetic variation influences DNA methylation. SNPs in distant LD also influenced DNA methylation at a different probe (cg08031982) in the fetal brain. Further evidence comes from published studies in lung, breast, and kidney tissues,31 as well as in utero,50 all of which have shown that the genetic variation at the same 16q24.2 region influences methylation at the cg08031982 probe. Interestingly, the CpG sites influenced by the locus appear to differ by tissue, with different probes affected in whole blood compared to fetal brain. This might imply tissue-specific functional consequences of the locus and therefore highlights the importance of performing follow-up experiments in appropriate tissues. Based on the evidence presented here, we can only speculate on how genetic variation at the locus leads to increased risk of SVS. One hypothesis is that expression of ZCCHC14, or other proteins, is mediated through altered methylation of the probes identified. This might occur, in part, in response to environmental stimuli. Evaluating these hypotheses in a relevant tissue type will be an important future analysis to identify the causal mechanisms leading to SVS.
This study has limitations. Our results suggested that the association may be present in other ethnicities, but we had an insufficient number of cases to establish common risk conclusively. Follow-up studies are therefore required in other ethnic groups. In addition, downstream functional experiments will be required to determine the consequences of the identified association. The mRNA expression and methylation analyses presented herein were constrained by available tissue types. Validation of the findings in more disease-relevant tissue types, such as cerebral small vessels, therefore represent important follow-up analyses, although obtaining such tissue in a state to allow mRNA studies is very challenging. We performed mRNA expression and methylation analyses using either the lead SNP (rs12445022) or three LD SNPs. The results should be interpreted with the limitation that we cannot be certain that any of these SNPs is the causal variant. Radiological confirmation of SVS in this study was performed using either computed tomography or MRI. Evidence shows that MRI is considerably more reliable at identifying SVS. Replication of the association in an MRI-confirmed population may therefore provide a more-accurate estimate of the effect of the locus on SVS risk. Similarly, interrogation of causative classification system definitions of SVS may provide further insights.51 Another method of interrogating the combined influence of age and genotype is by testing for an interaction. In this analysis, we were unable to do this because age was not available in some sets of controls (eg, WTCCC2).
In this large genome-wide meta-analysis using an age-at-onset informed approach, we have identified the first genome-wide significant locus that is associated solely with SVS. Our findings, which point to subtle changes in gene expression and DNA methylation influencing disease risk, show that strategies that account for different liability across disease-related covariates, such as age, can identify novel associations with disease.
Acknowledgment
Matthew Traylor is funded by the NIHR Biomedical Research Centre based at Guy's and St Thomas’ NHS Foundation Trust and King's College London. Hugh Markus is supported by an NIHR Senior Investigator award and his work is supported by NIHR Comprehensive Biomedical Research Unit funding awarded to Cambridge University Hospitals Trust. Cathryn Lewis receives salary support from the National Institute for Health Research (NIHR) Mental Health Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King's College London. Collection of the UK Young Lacunar Stroke DNA Study (DNA Lacunar) was primarily supported by the Wellcome Trust (WT072952) with additional support from the Stroke Association (TSA 2010/01). Genotyping of the DNA Lacunar samples was supported by a Stroke Association Grant (TSA 2013/01). Robin Lemmens is a senior clinical investigator of FWO Flanders. Martin Dichgans received funding from the DFG (CRC 1123, B3) and an EU Horizon 2020 grant (agreement No. 666881 SVDs@target). The TwinsUK study was funded, in part, by the European Research Council (ERC 250157), and from the TwinsUK resource, which receives support from the Wellcome Trust and the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy's and St Thomas’ NHS Foundation Trust in partnership with King's College London. SNP Genotyping was performed by The Wellcome Trust Sanger Institute and National Eye Institute through NIH/CIDR. The SiGN study was funded by a cooperative agreement grant from the US National Institute of Neurological Disorders and Stroke, National Institutes of Health (U01 NS069208).
Author Contributions
M.T., R.M., C.M.L., B.B.W., and H.S.M. conceived and designed the study. M.T., B.B.W., and H.S.M. drafted the manuscript. M.T. and K.B.H. drew the figures. M.T., R.M., M.A.N., I.C., F.R., P.S., D.S., M.A.H.-B., C.L.M.S., P.M.R., G.B., V.T., R.L., C.L., J.F.M., J.R., M.D., B.B.W., and H.S.M. contributed acquisition and analysis of METASTROKE data sets. H.X., L.H., M.F., C.J., J.F.M., B.D.M., S.J.K., M.D., B.B.W., J.J.-C., J.W.C., R.S., A.S., R.L., A.L., O.M., R.P.G., R.L.S., T.R., K.R., D.K.A., J.A.J., O.R.B., S.W.-S., J-M.L., M.T., S.S.R., P.D.B., S.L.P., Q.W., P.F.M., D.W., C.D.A., and J.R. contributed acquisition and analysis of the NINDS-SIGN data. J.R., D.W., C.L., and C.D.A. contributed acquisition and analysis of ICH data. G.C., S.D., L.J.L., S.S., J.C.B., and W.T.L., Jr., contributed acquisition and analysis of neuro-CHARGE data. I.Y., T.D.S., J.T.B., E.H., and J.M. contributed acquisition and analysis of DNA methylation data. M.T., H.S.M., and N.S.R. contributed acquisition and analysis of WMH data. G.T., K.S., U.T., and S.G. contributed acquisition and analysis of deCODE data.
Potential Conflicts of Interest
Authors whose affiliations are listed as deCODE/Amgen are employees of deCODE/ Amgen.

