

Journal list menu

 Issue
IssueJournal of Inherited Metabolic Disease: Volume 30, Issue 3
279-407June 2007
Export Citations
Download PDFs
Review
Therapeutic concepts in succinate semialdehyde dehydrogenase (SSADH; ALDH5a1) deficiency (γ-hydroxybutyric aciduria). Hypotheses evolved from 25 years of patient evaluation, studies in Aldh5a1−/− mice and characterization of γ-hydroxybutyric acid pharmacology
- Pages: 279-294
- First Published: 24 April 2007
Uric acid changes in urine and plasma: An effective tool in screening for purine inborn errors of metabolism and other pathological conditions
- Pages: 295-309
- First Published: 19 May 2007
Original Articles
Deprivation of pantothenic acid elicits a movement disorder and azoospermia in a mouse model of pantothenate kinase-associated neurodegeneration
- Pages: 310-317
- First Published: 12 April 2007
Regionally selective decreases in cerebral glucose metabolism in a mouse model of phenylketonuria
- Pages: 318-325
- First Published: 24 April 2007
Dietary long-chain polyunsaturated fatty acid supplementation in infants with phenylketonuria: a randomized controlled trial
- Pages: 326-332
- First Published: 12 April 2007
Mitochondrial disease: Needs and problems of children, their parents and family. A systematic review and pilot study into the need for information of parents during the diagnostic phase
- Pages: 333-340
- First Published: 11 May 2007
Carnitine transporter and holocarboxylase synthetase deficiencies in The Faroe Islands
- Pages: 341-349
- First Published: 06 April 2007
A novel starch for the treatment of glycogen storage diseases
- Pages: 350-357
- First Published: 19 May 2007
High incidence of autoantibodies in Fabry disease patients
- Pages: 365-369
- First Published: 24 April 2007
Effects of cholesterol and simvastatin treatment in patients with Smith–Lemli–Opitz syndrome (SLOS)
- Pages: 375-387
- First Published: 11 May 2007
Statin therapy depresses total body fat oxidation in the absence of genetic limitations to fat oxidation
- Pages: 388-399
- First Published: 05 April 2007
Short Report
A trial with N-carbamylglutamate may not detect all patients with NAGS deficiency and neonatal onset
- Page: 400
- First Published: 09 May 2007
Intermediate hyperhomocysteinaemia and compound heterozygosity for the common variant c.677C>T and a MTHFR gene mutation
- Page: 401
- First Published: 25 April 2007
Renal Fanconi syndrome with ultrastructural defects in lysinuric protein intolerance
- Pages: 402-403
- First Published: 19 May 2007
Summary
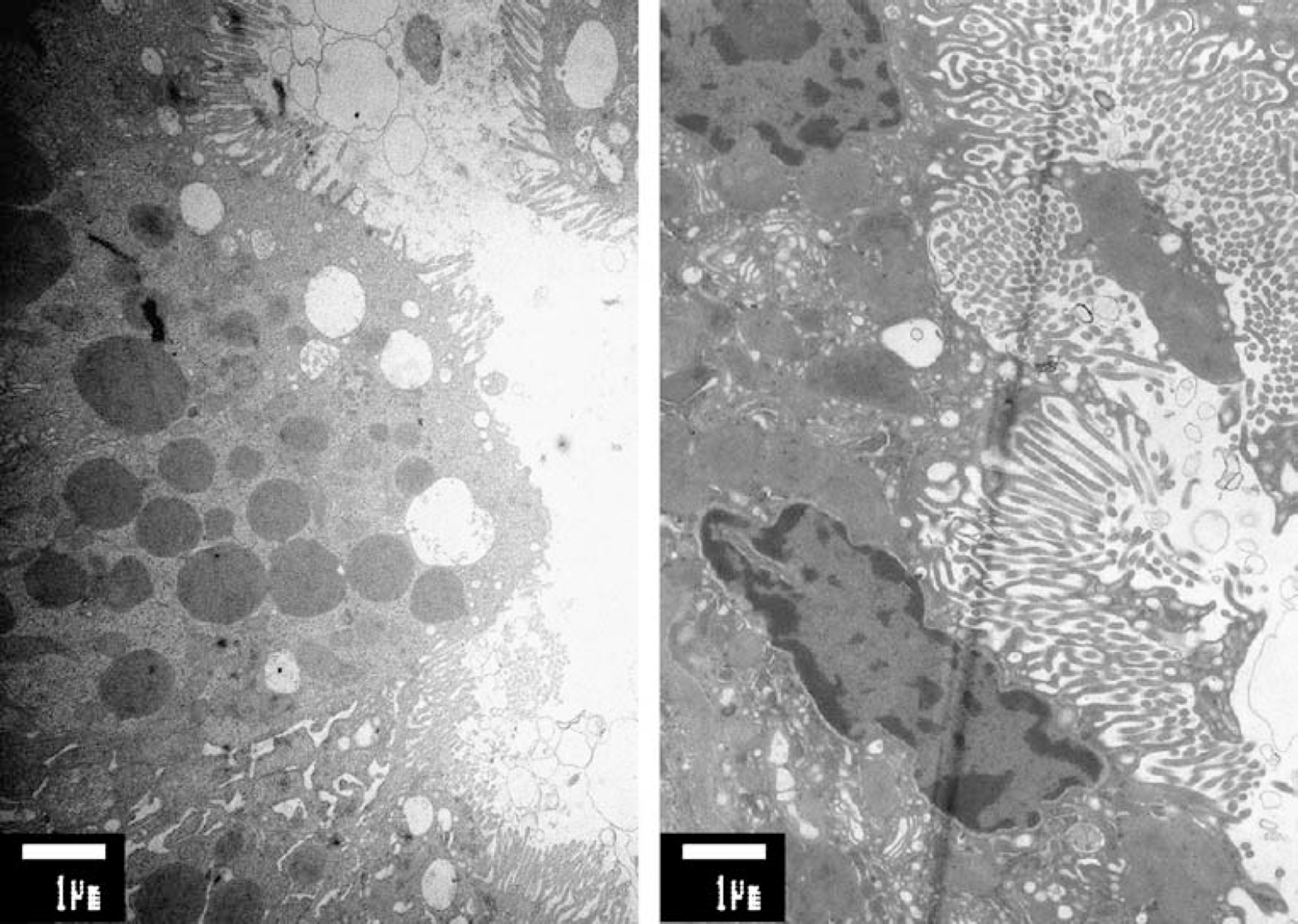
Renal Fanconi syndrome developed rapidly in a 3-year-old Moroccan girl with established lysinuric protein intolerance. She was hospitalized because of lowered consciousness, uncoordinated movements and hepatosplenomegaly after a febrile period. Laboratory investigations revealed plasma ammonia 270 μmol/L (normal <70 μmol/L), ferritin 159 μmol/L (normal 2–59 μmol/L), LDH 1180 U/L (normal 26–534 U/L). LPI was diagnosed based on the findings of reduced plasma ornithine, arginine and lysine, and an increased level of glutamine. Urinary orotic acid (645 μmol/mmol creatinine; normal <3.6) was strongly increased. A defect in the SLC7A7 amino acid transporter was established (homozygous c.726G>A mutation). Detailed renal function tests including an acid challenge test, bicarbonate loading, and tubular maximal reabsorption of glucose showed complex tubular dysfunction. No evidence of respiratory chain defects was found in muscle or kidney tissue. No morphological abnormalities were demonstrated in the mitochondria. Ultrastructural analysis of proximal tubular cells showed vacuolization and sloughing of the apical brush border (Fig. 1). Renal involvement in LPI has only been described in a few reports; however, no detailed studies of the renal acidification mechanism were performed. Our patient had evidence of a full-blown Fanconi syndrome. Surprisingly, a metabolic acidosis was found with a moderately increased serum anion gap combined with repeatedly normal plasma organic acid values. This finding is in contrast with the diagnosis of renal tubular acidosis. Patients with hyperlysinaemia have a similar heavy load on the renal tubules; they never develop a renal Fanconi syndrome. Therefore, we consider the intratubular accumulation of lysine an unlikely candidate for the development of the renal Fanconi syndrome.
1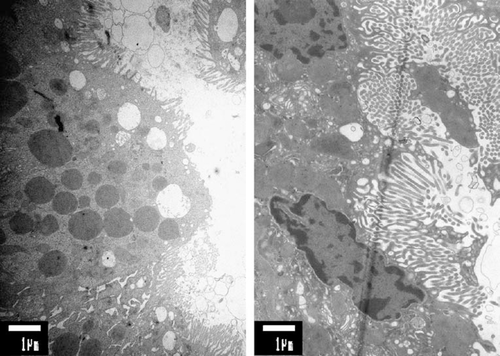
Left panel shows irregular and partially absent brush border in the patient with LPI. The panel on the right shows the regular brush border architecture in a patient of the same age (biopsy taken in remission phase of idiopathic nephritic syndrome). Note also that in this normal specimen the villi are longer than in the patient with LPI
Prenatal diagnosis by amniocentesis and chorionic villus biopsy of mtDNA mutation 8993T>G
- Page: 404
- First Published: 11 May 2007
Low citrulline may not be diagnostic of ornithine transcarbamylase deficiency: A case report
- Page: 405
- First Published: 03 April 2007
Effects of a fat load and exercise on asymptomatic VLCAD deficiency
- Page: 405
- First Published: 24 April 2007
Serum lipid and lipoprotein profile of patients with glycogen storage disease types I, III and IX
- Page: 406
- First Published: 03 April 2007
Transferrin hypoglycosylation in hereditary fructose intolerance: Using the clues and avoiding the pitfalls
- Page: 407
- First Published: 24 April 2007
Sign up for email alerts
Tools



