


All Ecology and Evolution Articles
Export Citations
Download PDFs
-
Mitochondrial genomes of four slug moths (Lepidoptera, Limacodidae): Genome description and phylogenetic implicationsoa
Graphical Abstract
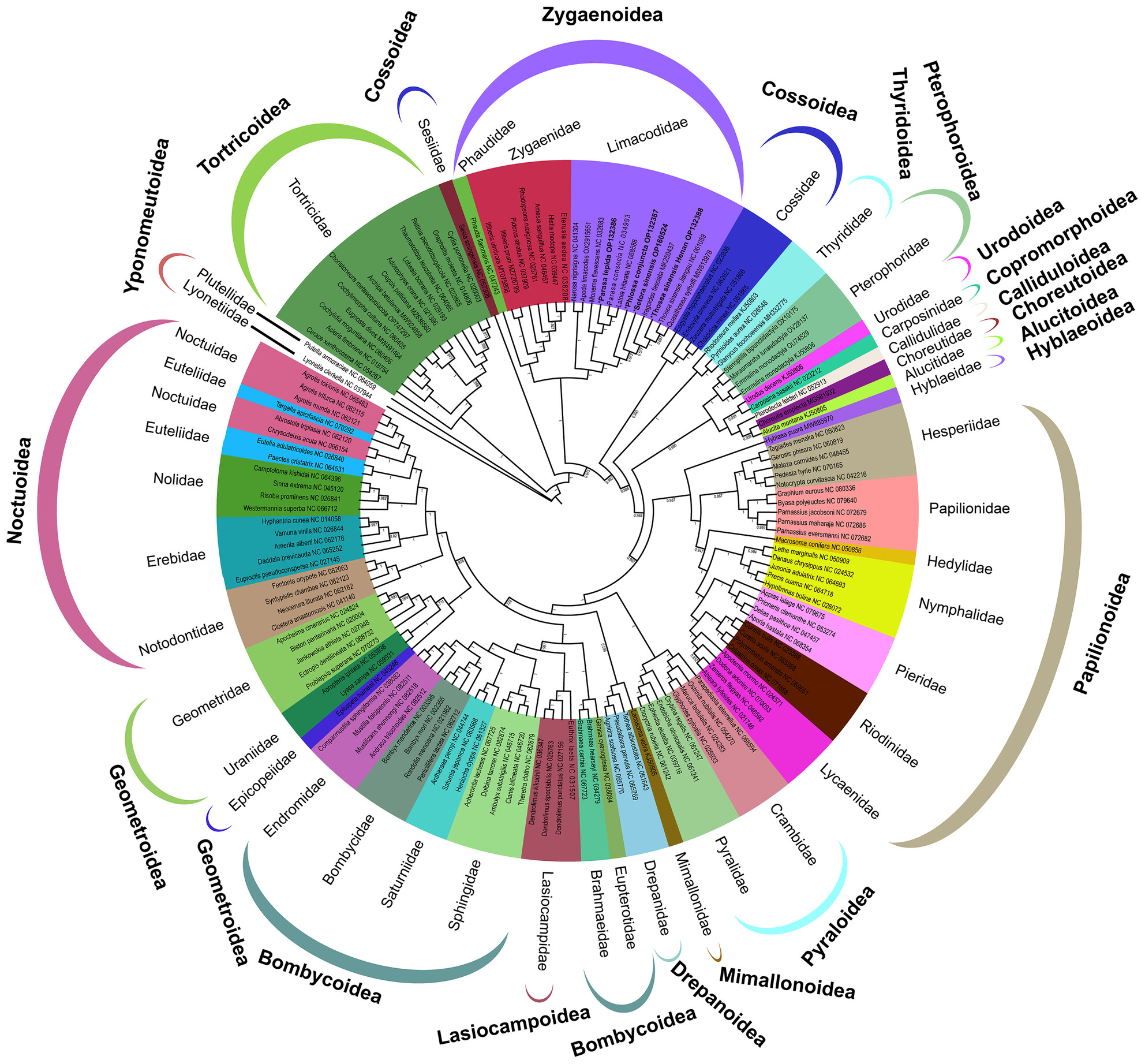
In this study, we sequenced the mitochondrial genomes of Parasa lepida, Phlossa conjuncta, Thosea sinensis, and Setora sinensis, and performed the first comparative genomic analyses of Limacodidae. The use of the ATT start codon by the cox1 gene in P. lepida is the first documented use of this start codon in Limacodidae mitogenomes. Within Zygaenoidea, Limacodidae was recovered as monophyletic, and the phylogenetic relationships were recovered as (Phaudidae + Zyganidae) + Limacodidae in all six phylogenetic trees.
-
Characterization of the complete mitochondrial genome of an endemic species in China, Aulocera merlina (Lepidoptera: Nymphalidae: Satyrinae) and phylogenetic analysis within Satyrinaeoa
Graphical Abstract
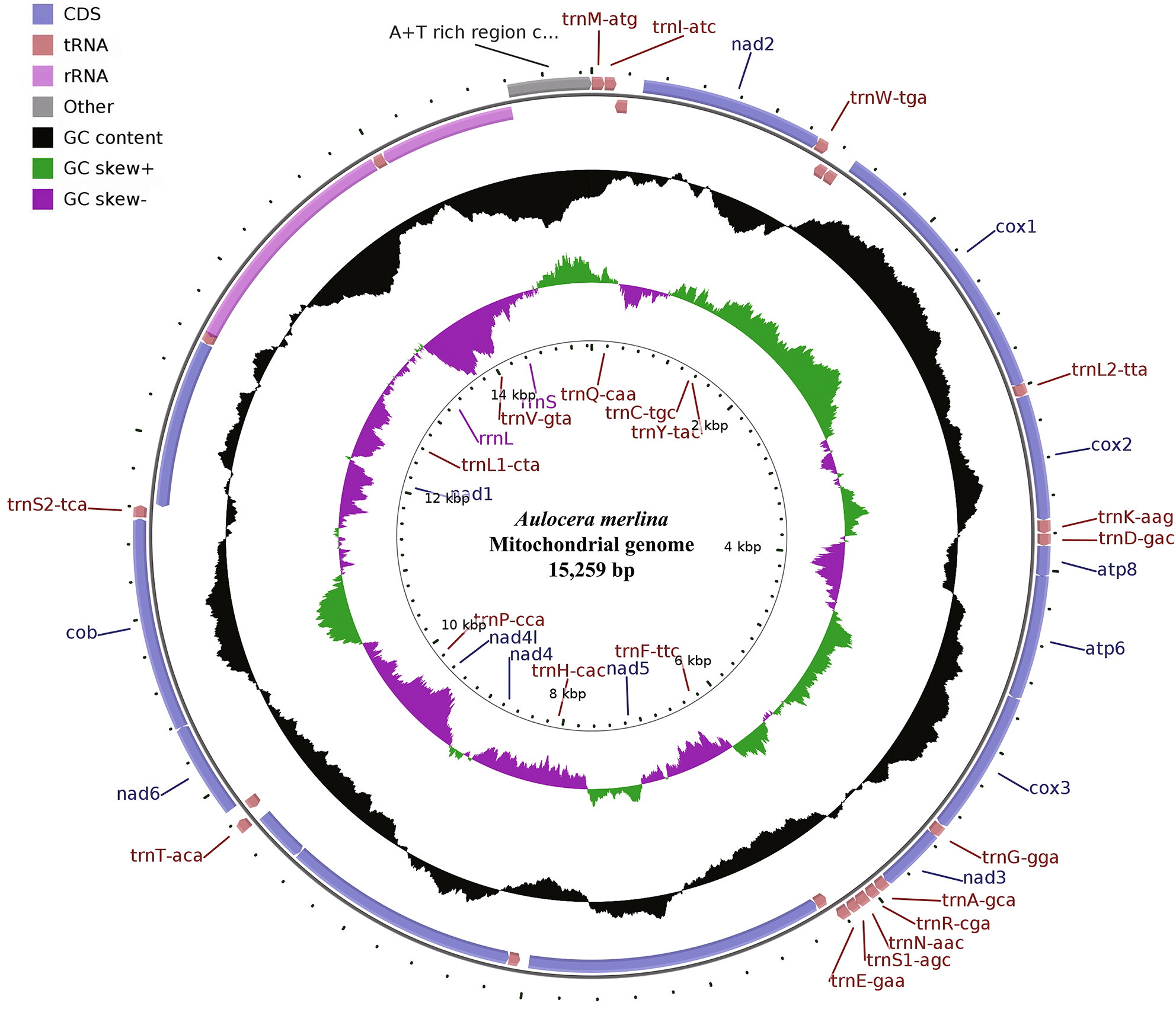
In this paper, the complete mitochondrial genome sequence of an endemic species in China, Aulocera merlina (Lepidoptera: Nymphalidae: Satyrinae) was determined and compared with that of other known mitogenomes of Satyrinae species. Moreover, the phylogenetic trees were reconstructed based on the available mitogenome sequences, including the newly sequenced mitogenome, to gain a better understanding of the phylogenetic relationships among the major lineages of the Satyrinae.
-
Genome sequencing, comparative analysis, and gene expression responses of cytochrome P450 genes in Oryzias curvinotus provide insights into environmental adaptationoa
Graphical Abstract
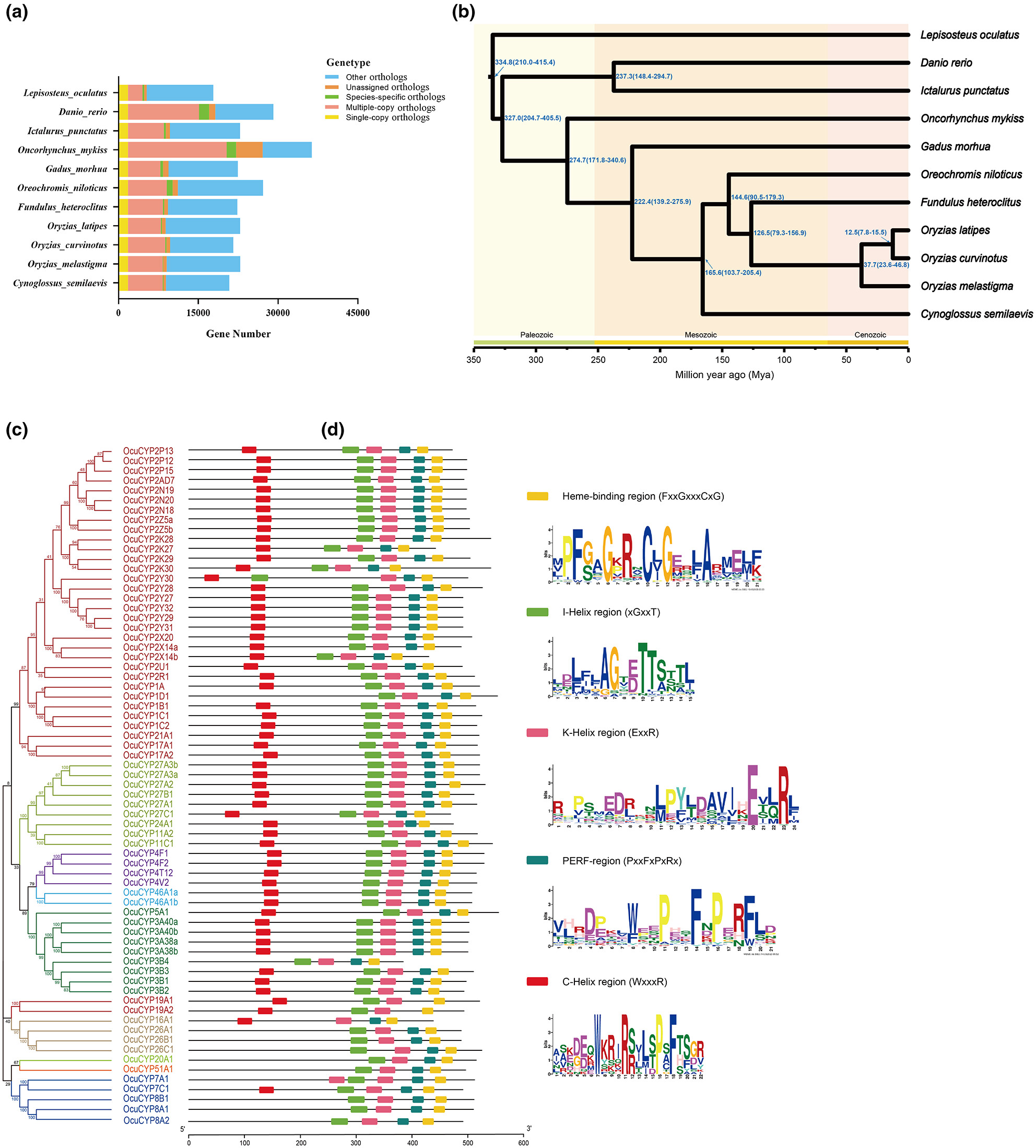
This chromosome-level genomic resource provides crucial biological insights to elucidate the functional roles of expanded CYPs in environmental adaptation, sexual development, early life history, and conservation in the anthropogenically impacted mangrove habitats of O. curvinotus.
-
Chloroplast genome-based genetic resources via genome skimming for the subalpine forests of Japan and adjacent regionsoa
Graphical Abstract
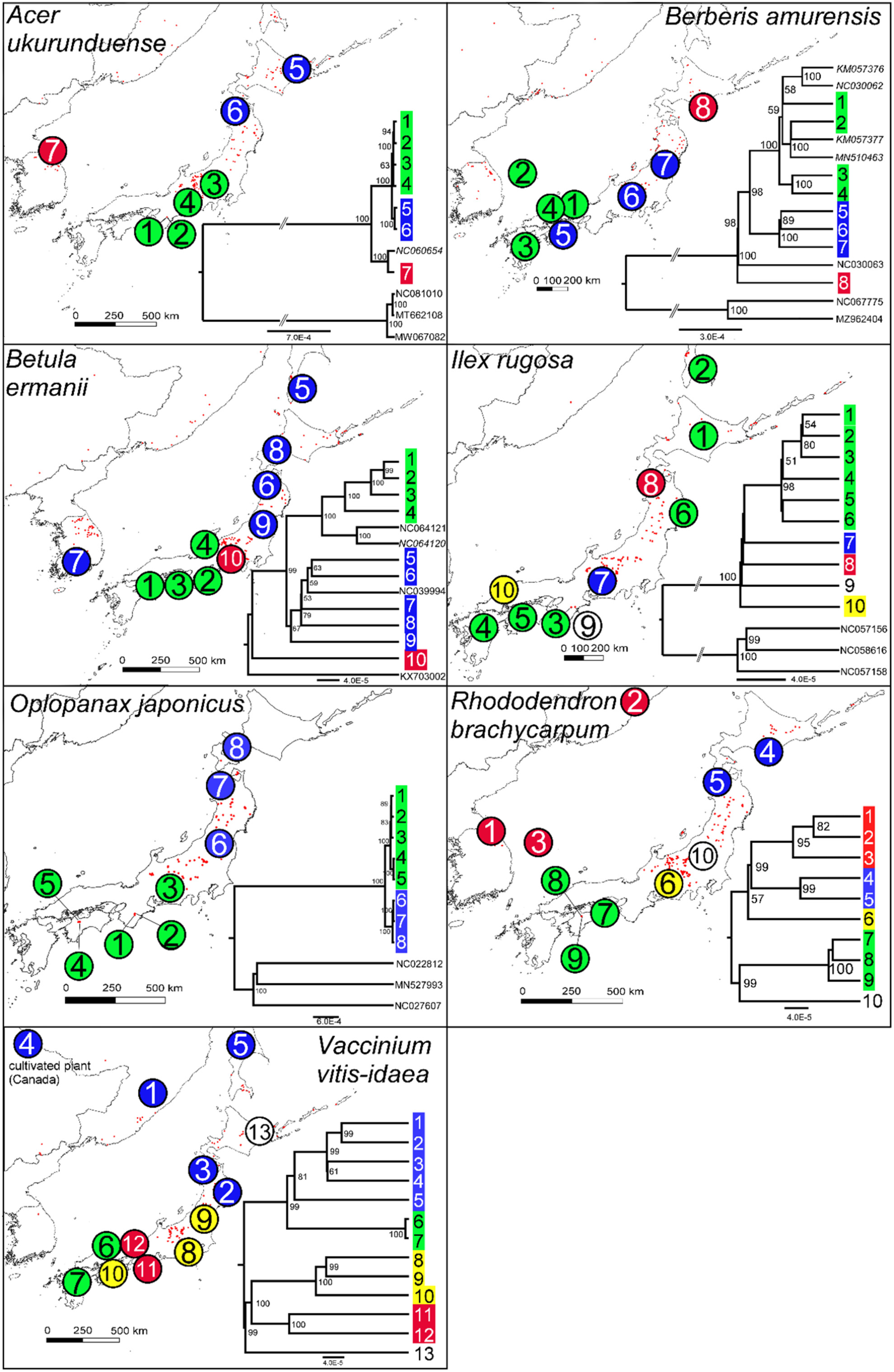
In this study, we assemble 92 whole chloroplast genomes from 12 subalpine plant species (5 conifers and 7 angiosperms) and identify intraspecific variation and lineages that will be useful for evolutionary, phylogeographic and conservation-focused studies of the threatened subalpine forest biome of Japan and adjacent regions in northeast Asia.
-
Development of a highly polymorphic chloroplast SSR set in Abies grandis with transferability to other conifer species—A promising toolkit for gene flow investigationsoa
Graphical Abstract
We introduce a remarkably polymorphic SSR marker set for various Abies species, which can be useful for other conifer genera, such as Cedrus, Pinus, Pseudotsuga or Picea. In total, 17 cpSSRs showed reliable amplification and polymorphisms in Abies grandis with a total of 68 haplotypes detected. All 17 cpSSRs amplified in the tested Abies spp. In the other tested species, except for Taxus baccata, at least one primer was polymorphic.
-
Insights into phylogenetic positions and distribution patterns: Complete mitogenomes of two sympatric Asian horned toads in Boulenophrys (Anura: Megophryidae)oa
Graphical Abstract
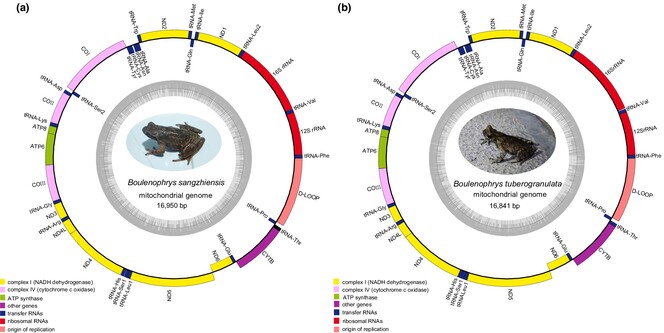
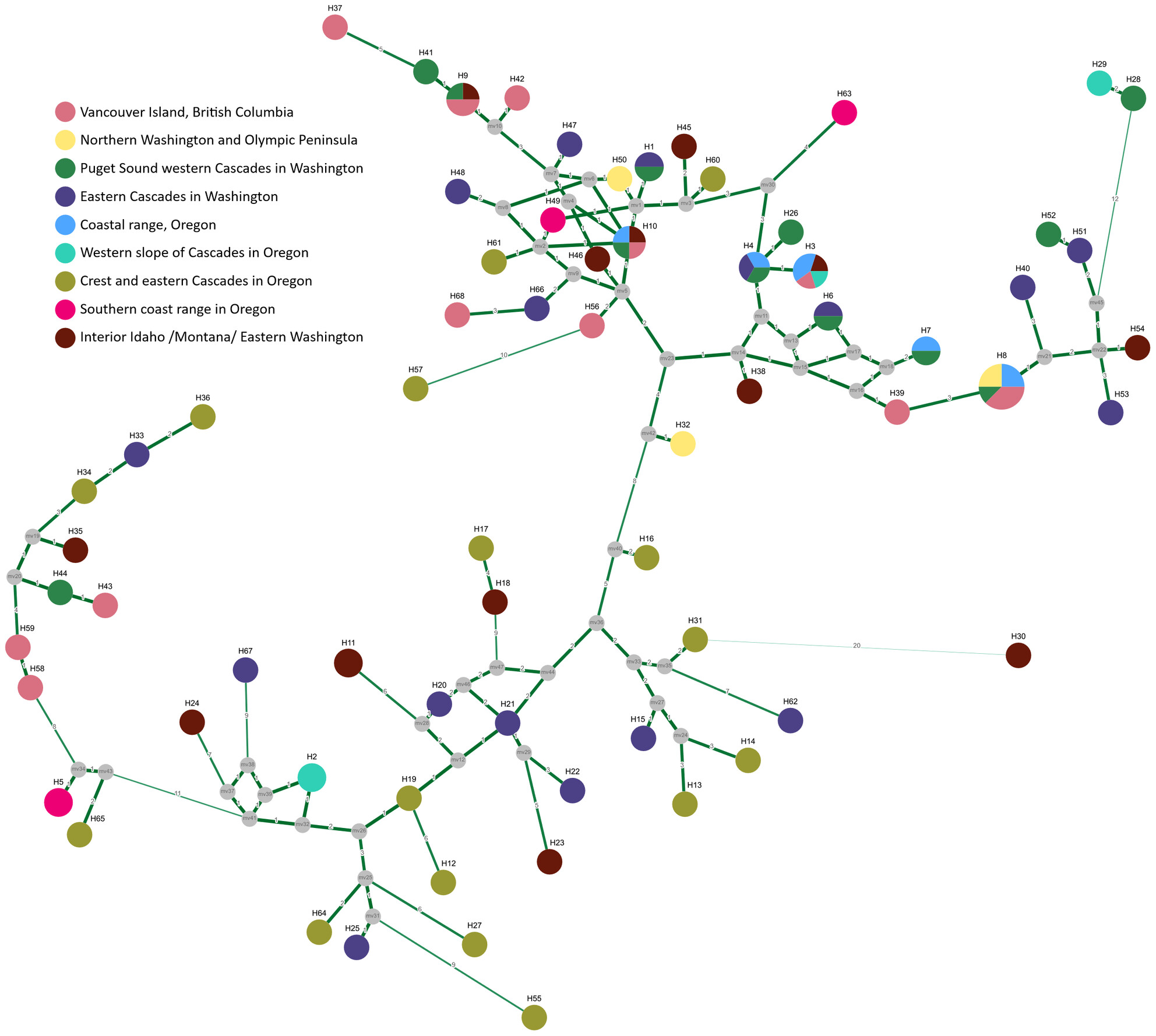
The mitogenomes of two endemic and threatened Asian horned toads, Boulenophrys sangzhiensis and Boulenophrys tuberogranulata, were sequenced and reported for the first time. A phylogenetic reconstruction based on all available mitogenomes within Megophryidae revealed a very special phylogenetic and distributional pattern of the two sympatric species relative to other congeners in Boulenophrys.
-
The complete chloroplast genome of Marupa (Simarouba amara Aubl., Simaroubaceae)oa
Graphical Abstract
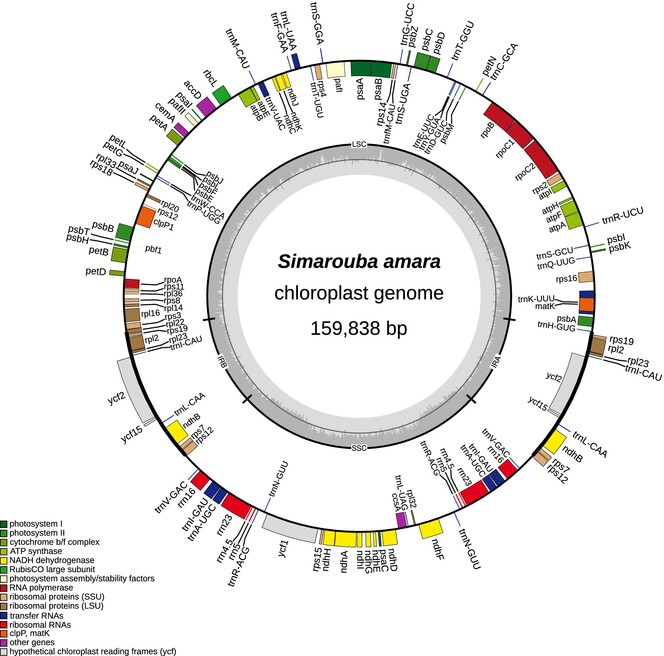
We provide the first plastome sequence of Marupa (Simarouba amara Aubl., Simaroubaceae). Sequence was generated using Oxford Nanopore Technology. The chloroplast is 159,838 bp, includes 131 genes in total and presents a classic quadripartite structure.
-
De-novo assembly of four rail (Aves: Rallidae) genomes: A resource for comparative genomicsoa
Graphical Abstract
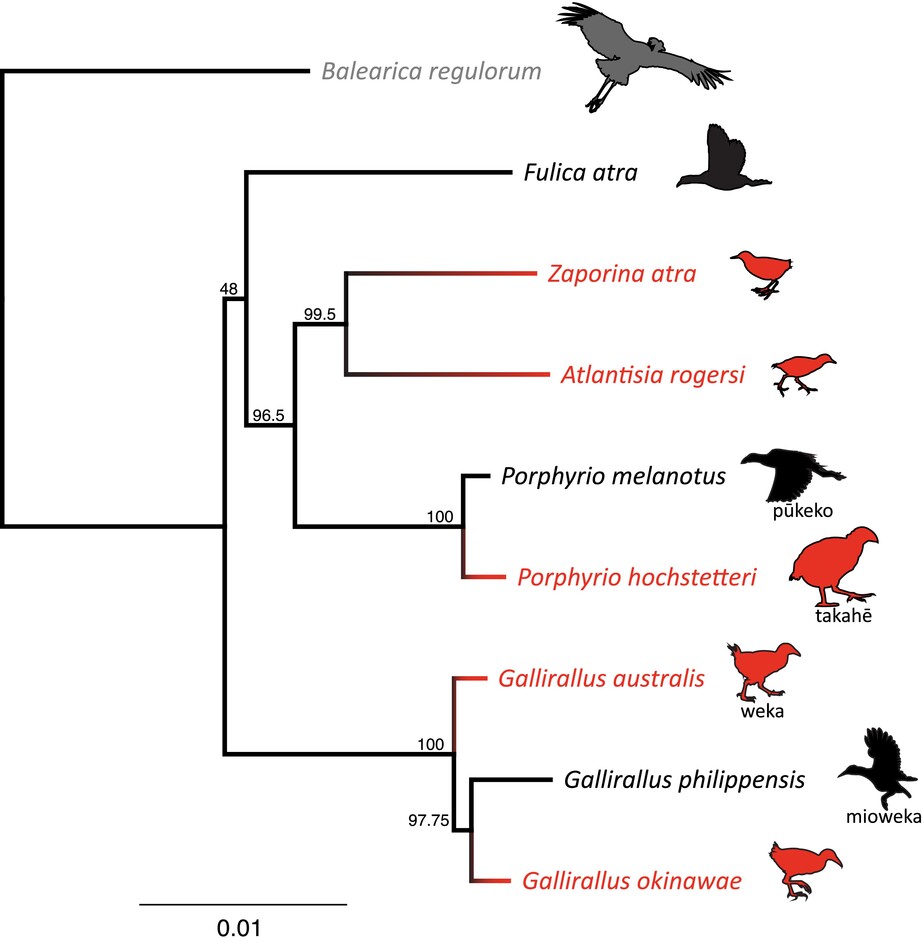
We report new annotated genome assemblies for four New Zealand rails: two native volant species, and two endemic flightless species. This study significantly increases the number of available rallid genomes and will enable future comparative genomic studies on the evolution of this family. We find our genome assemblies and annotations are comparable to similar species. Heterozygosity was high in the two volant species and low in the two endemic flightless species.
-
A reproducible workflow for assembling the mitochondrial genome of Acheta domesticus (Orthoptera: Gryllidae)oa
Graphical Abstract
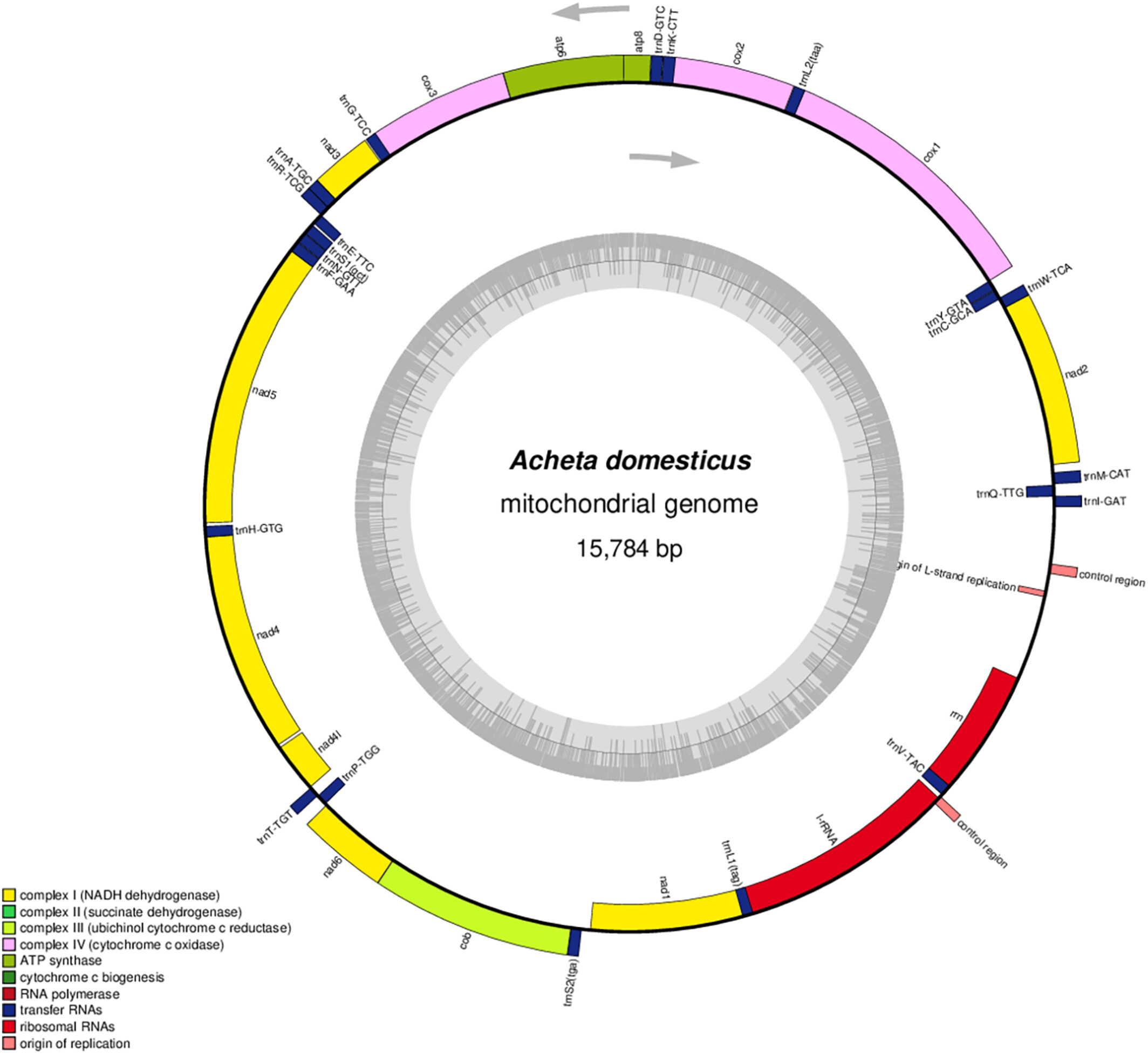
This study presents the comprehensive assembly and analysis of the complete mitochondrial genome of the economically important house cricket, Acheta domesticus. The high-quality mitogenomic data obtained through our robust bioinformatic workflow serve as an invaluable resource for precise DNA barcoding, species identification, and evolutionary studies within the order Orthoptera. The comparative genomic analysis and phylogenetic insights derived from this research shed light on the evolutionary relationships and genetic diversity of A. domesticus and related cricket species.
-
DNA barcoding of marine rocky reef fishes from northern Peru suggests a parapatric speciation in the Tropical Eastern Pacificoa
Graphical Abstract
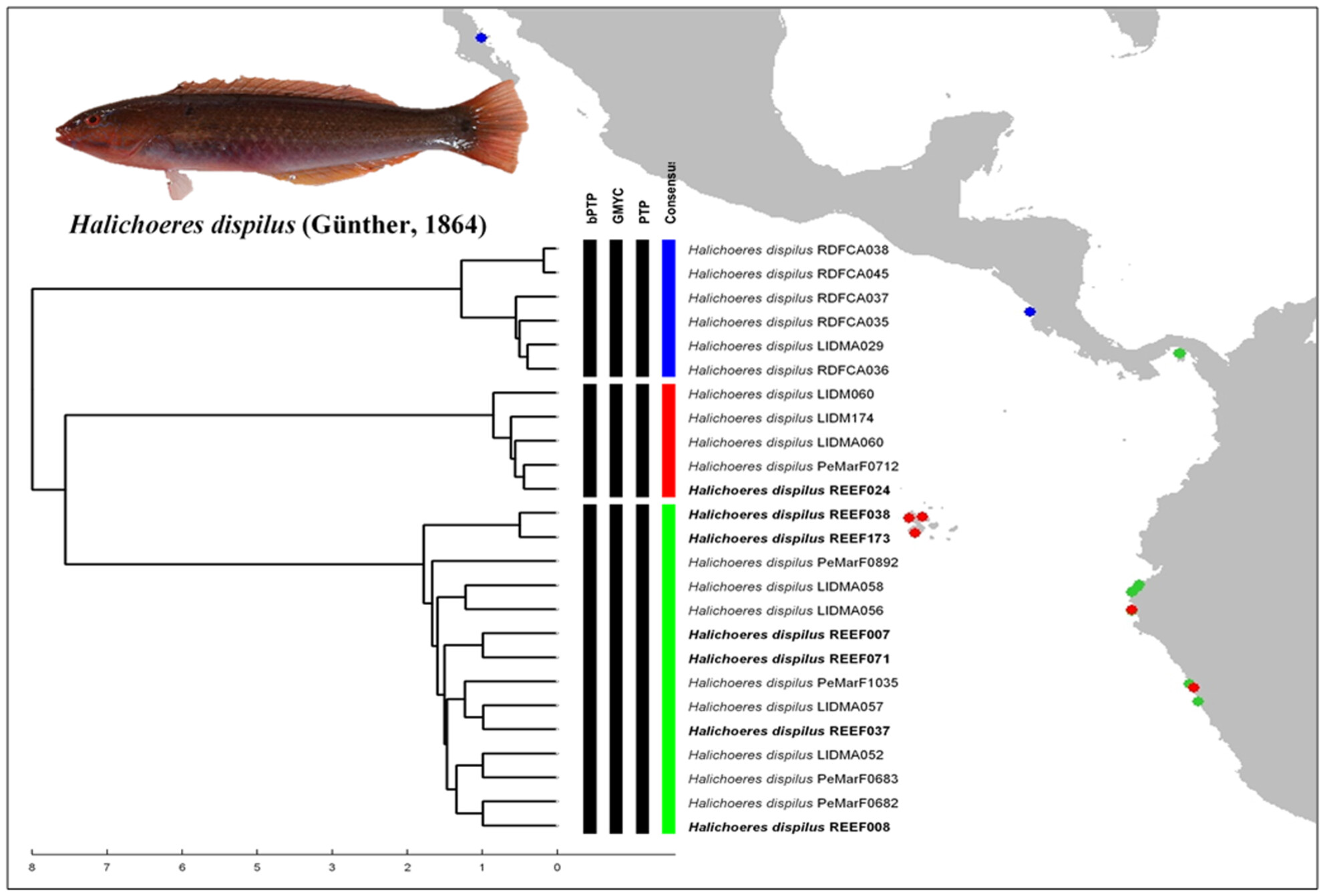
Using a DNA barcoding technique we found evidence that reinforce the hypotheses of how parapatric speciation could drive the origin of new species in the TEP.
-
Comparative mitogenomics of marine angelfishes (F: Pomacanthidae)oa
Graphical Abstract
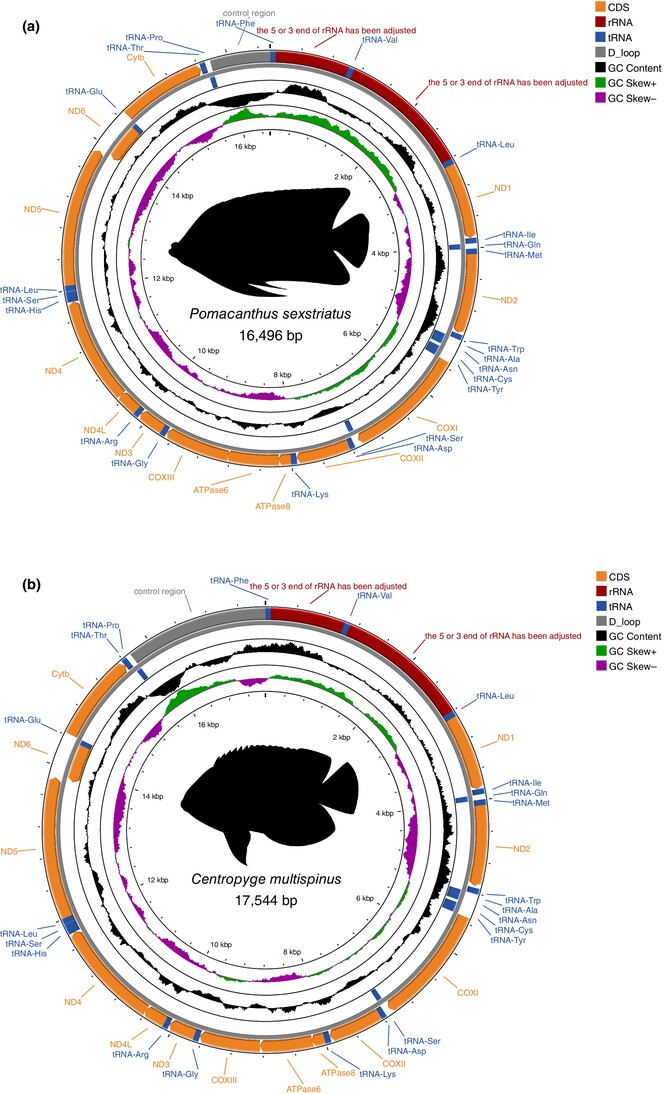
This study describes newly assembled and annotated mitogenomes harvested from off-target reads of ultraconserved element captured data for six pomacanthid species. Secondly, it investigates evolutionary patterns of protein-coding genes across all mitogenomes available for the family on the NCBI Reference Sequence Database. Finally, we generated the most comprehensive phylogenetic tree for the family using complete reference mitogenomes.
-
Rampant intraspecific variation of plastid genomes in Gentiana section Chondrophyllaeoa
Graphical Abstract
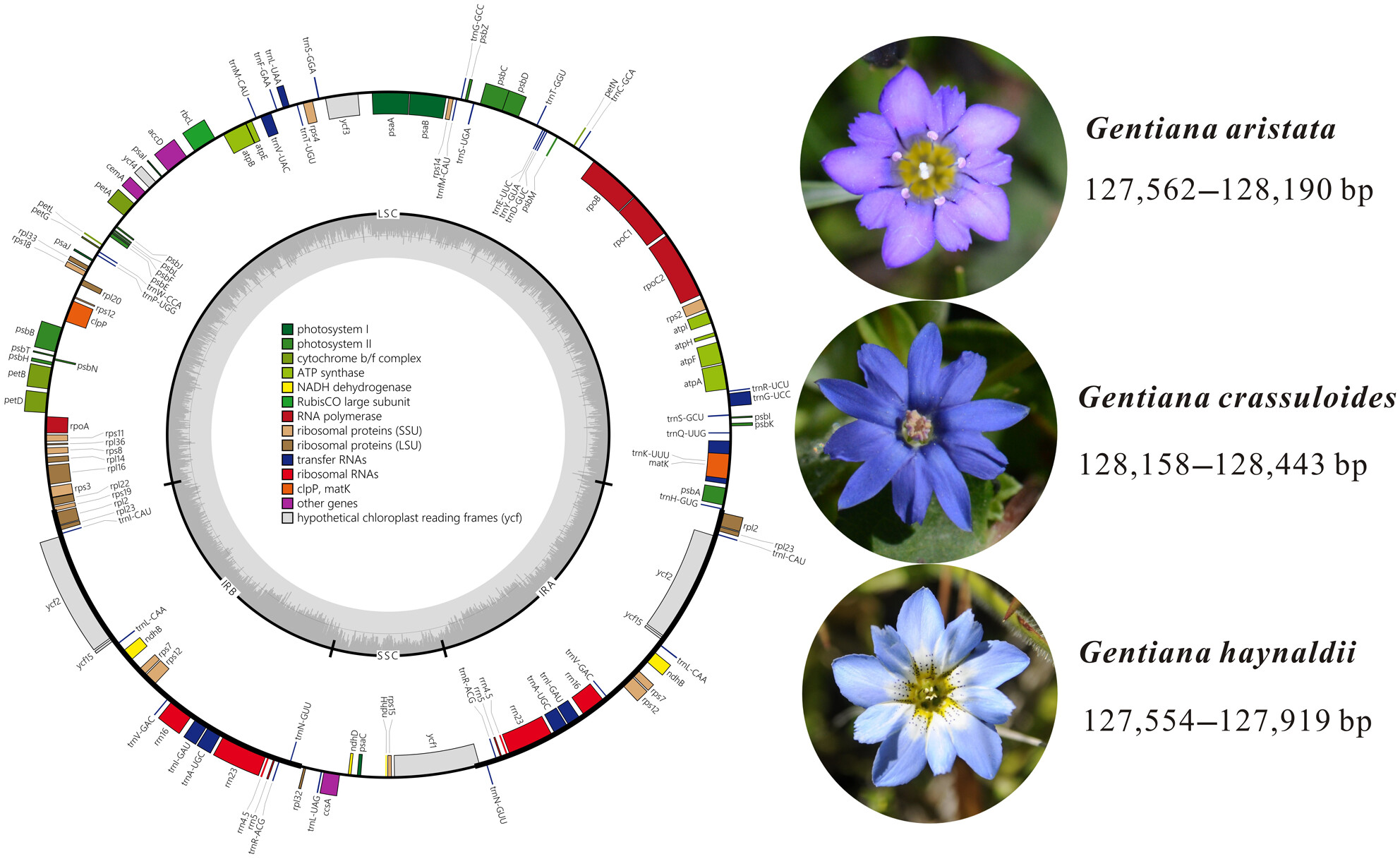
Significant high level of intraspecific plastome size variation and genetic diversity are detected in three annual gentians of Gentiana section Chondrophyllae s.l.
-
Mitochondrial genomes of Macropsini (Hemiptera: Cicadellidae: Eurymelinae): Structural features, codon usage patterns, and phylogenetic implicationsoa
Graphical Abstract

The aim of this study is to address the patterns and processes that explain the structure and the evolution of the mitogenomes of Macropsini, while contributing to the resolution of systematic issues involving five of their genera. To this task, the mitogenomes of 26 species of the tribe were sequenced and characterized, and their phylogenetic relationships were reconstructed.
-
Optimising recovery of DNA from minimally invasive sampling methods: Efficacy of buccal swabs, preservation strategy and DNA extraction approaches for amphibian studiesoa
Graphical Abstract
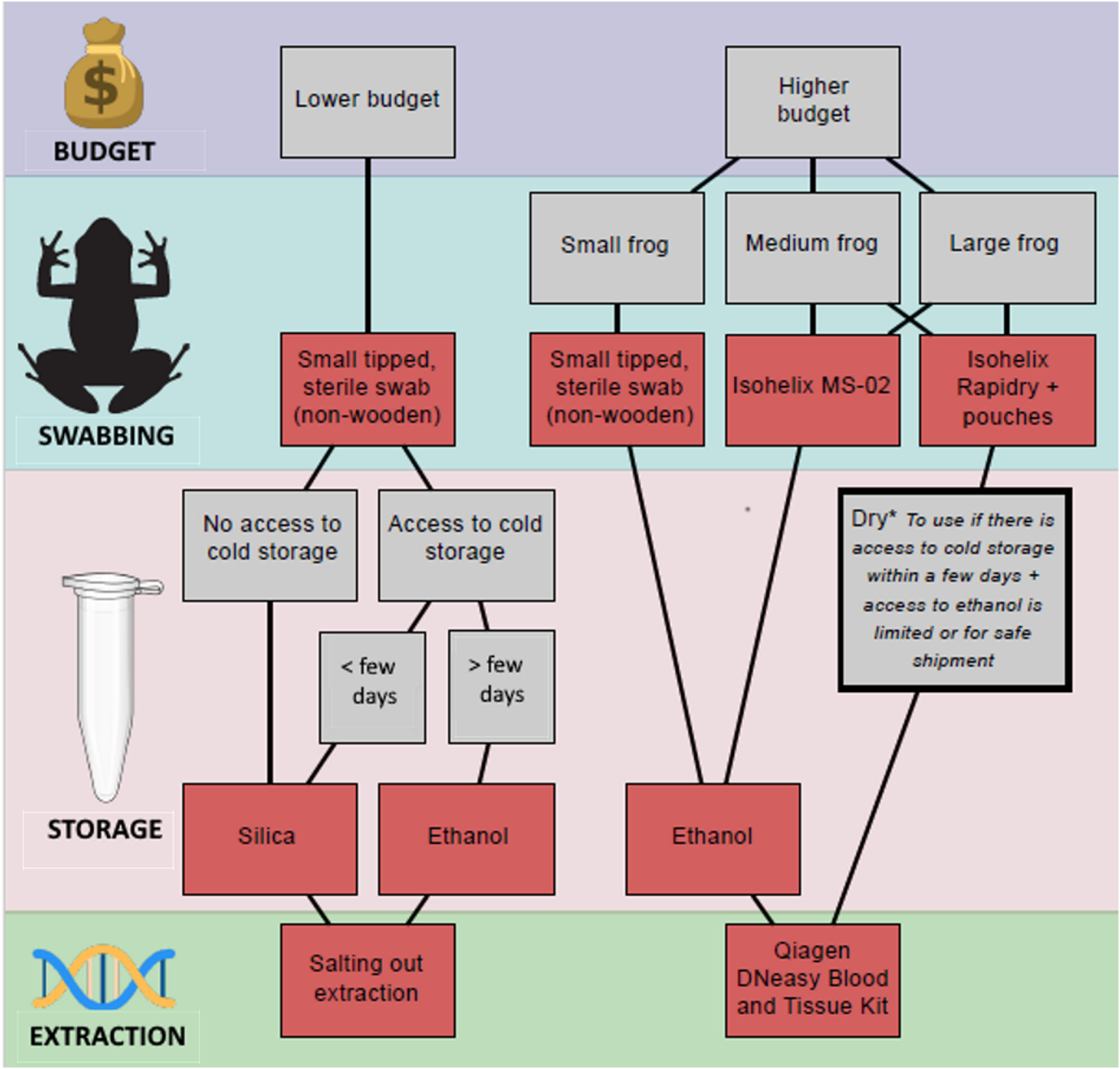
In this multiple-case study, we tested a comprehensive set of factors impacting DNA recovery from buccal swabs, including the type of swab, storage type and conditions and DNA extraction method. Based on our results, we are able to propose recommendations for improved DNA recovery, taking into consideration budget and field condition constraints.
-
DNA barcoding and cryptic diversity in fishes from the Ili River Valley in China, Xinjiangoa
Graphical Abstract
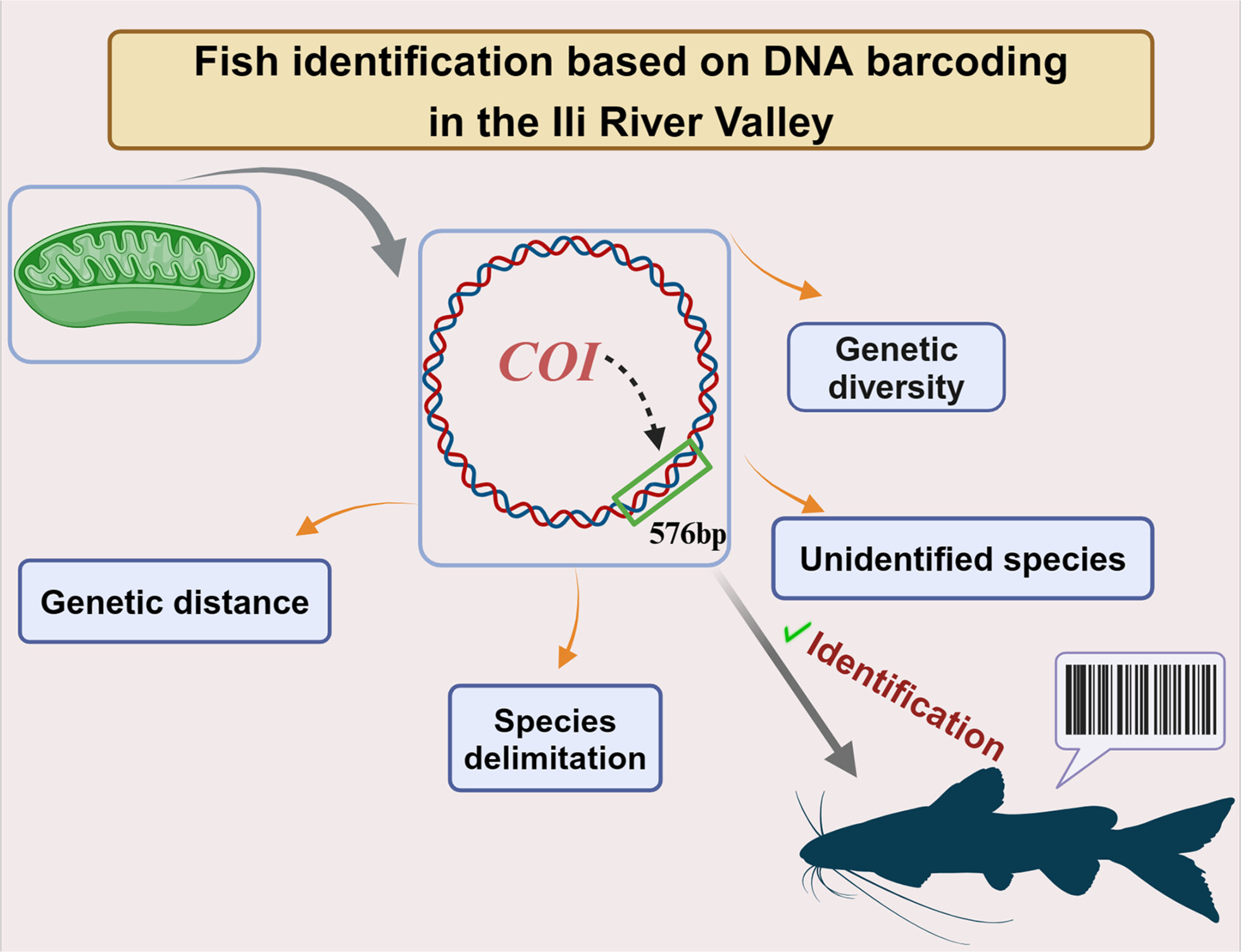
With a 95% success rate in identifying species, DNA barcoding based on the mitochondrial COI gene was validated in this study using genetic diversity, species delimitation, genetic distances, and unidentified species analysis. Simultaneously, we discovered that the Ili River Valley's fish species populations are drastically decreasing to the point where the area faces a major ecological risk.
-
Comparative study on chloroplast genome of Tamarix speciesoa
Graphical Abstract
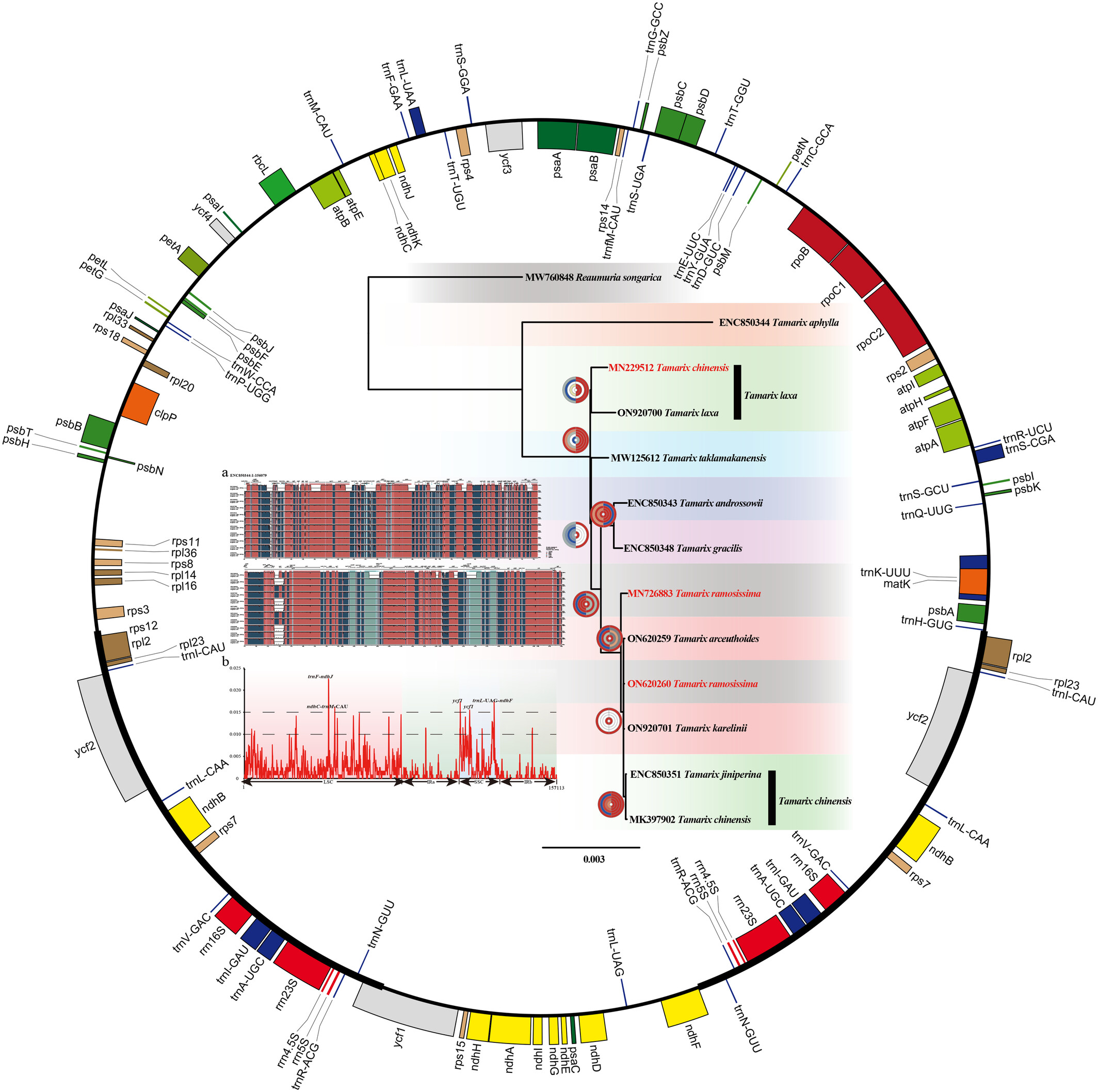
Phylogenetic relationship of Tamarix.
-
Chromosome-Level Genome Assembly for the Chinese Serow (Capricornis milneedwardsii) Provides Insights Into Its Taxonomic Status and Evolutionoa
Graphical Abstract
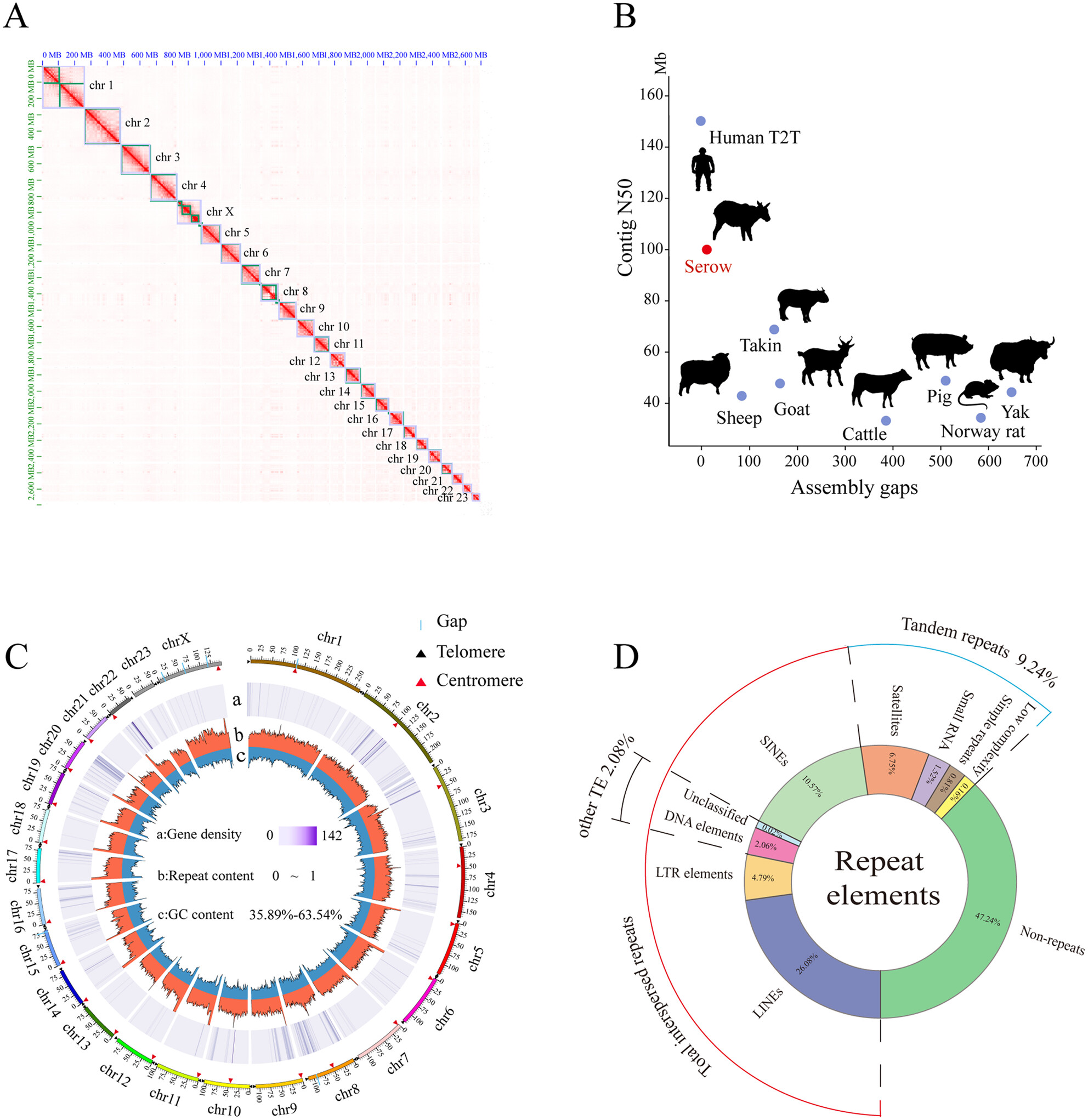
We constructed a high-quality chromosome-level reference genome of C. milneedwardsii for the first time. The Chinese serow was more closely related to muskox, and the karyotype of serow (2n = 48) occurred six chromosome fusions. Interestingly, compared to other Caprinae species, the MYH6 protein of Chinese serow occurred two mutations (E1520S and G1521S) which were conserved in Cetaceans.
-
The Complete Mitochondrial Genomes of Five Nivanini Species (Hemiptera: Cicadellidae: Evacanthinae) With Phylogenetic Analysisoa
Graphical Abstract
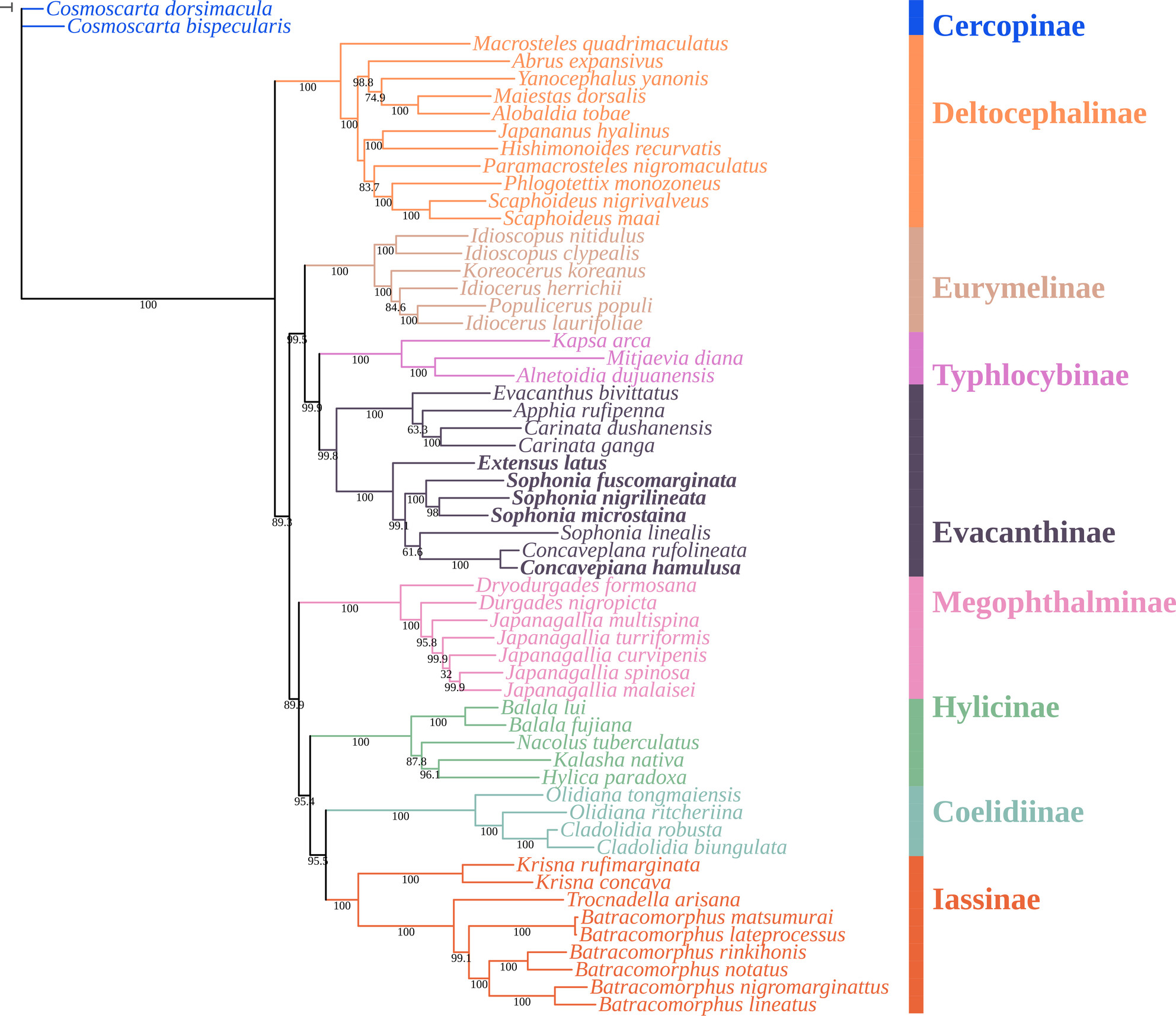
We conducted a comprehensive analysis by sequencing the mitogenomes of five Nivanini species and integrating this data with previously obtained sequencing results. Our investigation focused on exploring the characteristics of mitogenomes and establishing phylogenetic relationships.
-
Comprehensive Genomic Dataset of Chinese Lizardtail Herb and Comparative Genomic Analysis Provide Insights Into Its Paleo-Polyploidization Eventoa
Graphical Abstract
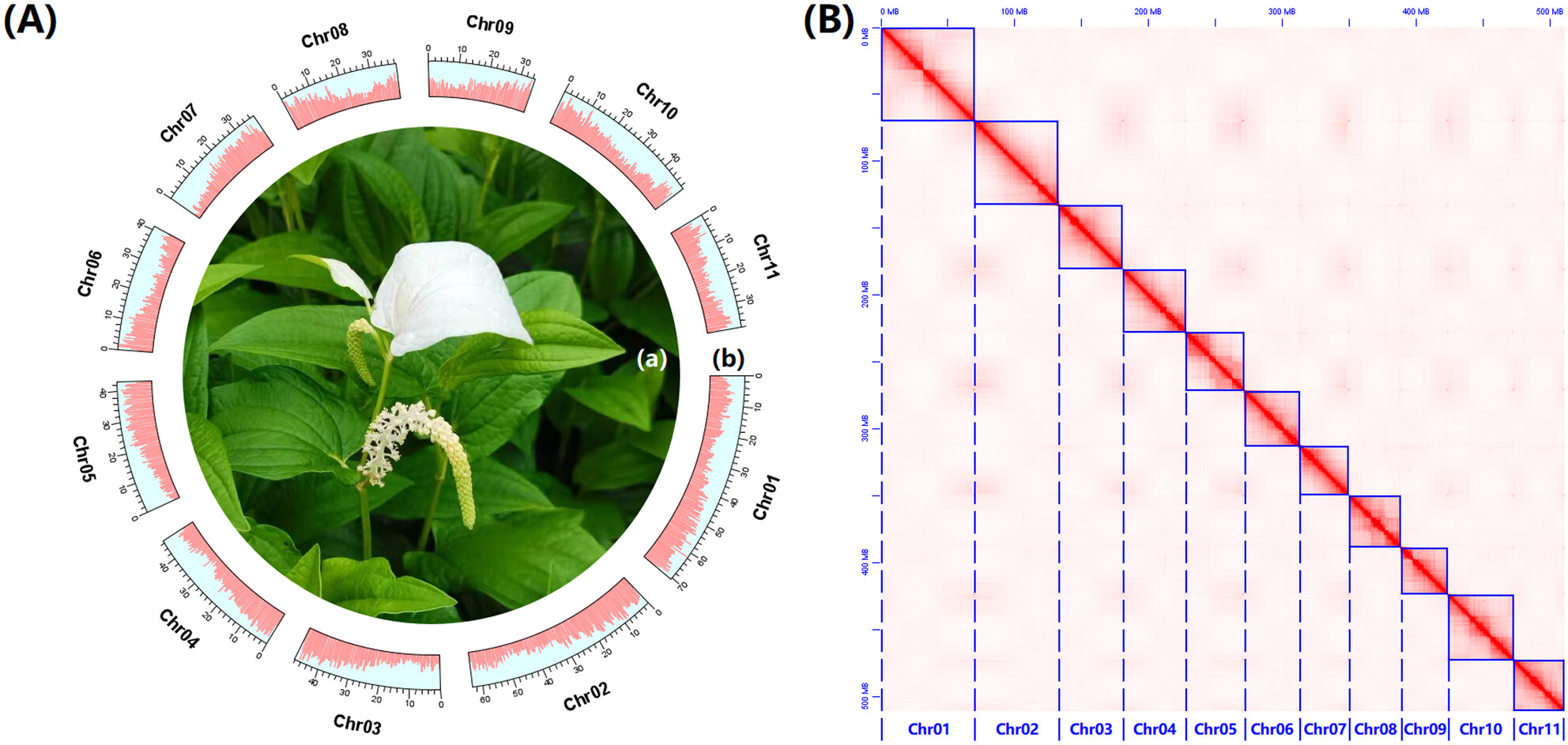
In this study, we provided a comprehensive genomic dataset for S. chinensis. Additionally, we observed that the S. chinensis genome underwent a paleo-tetraploidization event, followed by several chromosome fusion events, resulting in an aneuploid paleo-tetraploid state in its current genomic structure. Furthermore, we also observed that this paleo-tetraploidization event facilitated an expansion of the PEL family within the S. chinensis genome.