

QUIDDICH: QUick IDentification of DIagnostic CHaracters
Abstract
With the advent of molecular genetic methods, an increasing number of morphologically cryptic taxa has been discovered. The majority of them, however, remains formally undescribed and without a proper name although their importance in ecology and evolution is increasingly being acknowledged. Despite suggestions to complement traditional descriptions with genetic characters, the taxonomic community appears to be reluctant to adopt this proposition. As an incentive, we introduce QUIDDICH, a tool for the QUick IDentification of DIgnostic CHaracters, which automatically scans a DNA or amino acid alignment for those columns that allow to distinguish taxa and classifies them into four different types of diagnostic characters. QUIDDICH is a system-independent, fast and user-friendly tool that requires few manual steps and provides a comprehensive output, which can be included in formal taxonomic descriptions. Thus, cryptic taxa do not have to remain in taxonomic crypsis and, bearing a proper name, can readily be included in biodiversity assessments and ecological and evolutionary analyses. QUIDDICH can be obtained from the comprehensive R archive network (CRAN, https://cran.r-project.org/package=quiddich).
1 INTRODUCTION
In traditional taxonomy, taxa are usually described based on morphological and anatomical features (Jörger & Schrödl, 2013). However, since the advent of molecular genetic methods, in particular PCR, an exponentially increasing number of morphologically hard or impossible to distinguish animal and plant species, so-called cryptic species, has been discovered and gained recognition in ecology, evolutionary and conservation biology (Bickford et al., 2007; Struck et al., 2018). Unfortunately, the vast majority of cryptic species remains formally undescribed and without a proper name (Schlick-Steiner et al., 2007). It has been suggested to complement the formal descriptions of such morphologically cryptic species by including, for example, behavioral, ecological, biogeographic or, in particular, genetic data, that is, the very same data that often have led to their discovery. But although this pluralistic approach, which is referred to as integrative taxonomy (Dayrat, 2005; Padial, Miralles, De la Riva, & Vences, 2010; Schlick-Steiner et al., 2010), is frequently applied to identify taxa, the formal descriptions remain undone in probably the majority of cases. Botanists and zoologists appear to be particularly reluctant to include genetic data, although the nomenclatural codes are indifferent regarding the nature of data used in descriptions (International Commission on Zoological Nomenclature, 1999; Turland et al., 2018). In fact, Renner (2016) has found only 98 descriptions of species of plants, animals and fungi containing DNA data as of November 2015. In a fundamental paper, Jörger & Schrödl (2013, 2014) have provided an important starting point on how to deploy genetic data given in the form of alignments in which the columns represent the positional homology assumptions. To extract diagnostic characters, that is, those columns that are suitable to distinguish a taxon of interest from the remaining ones, they used the Characteristic Attribute Organization System (CAOS; Sarkar, Planet, & Desalle, 2008). Alternatively, the R package SPIDER (Brown et al., 2012) could be used for this purpose (Jörger & Schrödl 2014). As neither of the programs has been specifically designed for taxonomic applications, it is not surprising that one encounters problems in this particular context.
Characteristic attribute organization system provides a detailed output and distinguishes between different types of diagnostic characters (see below). However, it requires a fully resolved phylogenetic tree, which, for the purpose of sequence-based taxonomy, has to be rearranged so that the taxon of interest becomes outgroup to the remaining sequences (Jörger & Schrödl, 2013, 2014). This essentially wrong or definitely suboptimal tree serves as a guiding structure for the sequence comparisons conducted along the tree hierarchy. Eventually, the only taxon whose sequences are compared to all other sequences is the outgroup taxon. Hence, only for the latter do we get the comprehensive collection of diagnostic positions. If we are interested in a second taxon, we have to rearrange the tree again and rerun the analysis. A second issue concerns the so-called symplesiomorphy filter. Since one of CAOS’s original purposes is to classify a novel sequence into a tree, only apomorphic states are considered as useful information, whereas symplesiomorphic states are removed (Sarkar et al., 2002). The polarity, however, is tree-dependent. As the manipulated tree is arbitrary, the polarity of the character states is arbitrary as well. In addition, CAOS does not distinguish between gaps and masked alignment entries (Jörger & Schrödl, 2014). Both are considered as missing data, even though a masked entry indicates uncertainty about the true state, while a gap denotes an evolutionary event with a certain outcome, namely a deletion or insertion. A final disadvantage concerns the implementation in the software RubyCAOS (Sarkar et al., 2008), which is only available for Mac OS X 10.6 + and Linux. This system dependency limits its application. Additionally, as of November 2018, the Linux version produces an incomplete output as it is notably missing (among others) the CAOS groupFile.txt that is required to identify which part of the output belongs to the taxon of interest. Also, the online version of CAOS that was announced by Sarkar et al., (2008) is no longer available.
The alternative for CAOS that is currently available is the function nucDiag from the R package SPecies IDentity and Evolution in R (SPIDER) (Brown et al., 2012). On one side, 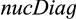 overcomes most of CAOS’s flaws making the application easier, faster and system- as well as phylogeny-independent (i.e., SPIDER does not require a guide tree). On the other side, its output is not as comprehensive, because it considers only two types of diagnostic characters (see below) and only returns the alignment positions of the identified characters without any information on the states that are characteristic for the taxon of interest. Additionally, it extracts diagnostic characters for every single taxon that is contained in the dataset. This may cause unnecessary computational costs if the user is just interested in one or a few taxa. The third disadvantage is again the treatment of gaps and masked alignment entries. Both of them are considered as “valid” character states, although at least the latter should definitely not be treated as such.
overcomes most of CAOS’s flaws making the application easier, faster and system- as well as phylogeny-independent (i.e., SPIDER does not require a guide tree). On the other side, its output is not as comprehensive, because it considers only two types of diagnostic characters (see below) and only returns the alignment positions of the identified characters without any information on the states that are characteristic for the taxon of interest. Additionally, it extracts diagnostic characters for every single taxon that is contained in the dataset. This may cause unnecessary computational costs if the user is just interested in one or a few taxa. The third disadvantage is again the treatment of gaps and masked alignment entries. Both of them are considered as “valid” character states, although at least the latter should definitely not be treated as such.
In order to provide a tool for the QUick IDentification of DIagnostic CHaracters, which overcomes the drawbacks while at the same time preserving the useful conceptual aspects of existing software, we developed the R package QUIDDICH. QUIDDICH is system-independent, easy to implement, fast, and produces a detailed output. Extending the concepts of CAOS and SPIDER, it can also deliver pairwise diagnostic characters, that is, characters that are suitable to distinguish pairs of taxa (Zielske & Haase, 2015). As genetic data have identified also higher cryptic taxa, for example Ecdysozoa and Lophotrochozoa, the subclades of Protostomia (Aguinaldo et al., 1997; Philippe, Lartillot, & Brinkmann, 2005), which may be more robustly analyzed on the protein level, we implemented functions that can search through both DNA and amino acid alignments. We hope that with an appropriate tool at hand taxonomists will no longer hesitate to include genetic data in descriptions and diagnoses of morphologically or otherwise hard or impossible to define taxa. Thus, cryptic taxa do not have to remain in taxonomic crypsis (Schlick-Steiner et al., 2007) and, bearing a proper name, can readily be included in biodiversity assessments and ecological and evolutionary analyses.
2 DATA INPUT AND OUTPUT
QUIDDICH requires an alignment (either nucleotides or amino acids) as well as a taxon vector whose i-th entry is the name of the taxon that the i-th row belongs to. Alignments may contain the IUPAC codes for the bases (resp. amino acids), – for gaps, and N (resp. X) for missing or ambiguous states or parts of the alignment that are to be masked, for example, in case of alignment ambiguities. The alignment must be stored using the classes DNAbin (nucleotides) or AAbin (amino acids) of the APE package (Paradis & Schliep, 2018). Fasta files can be imported and converted using adegenet's function fasta2DNAbin (Jombart, 2008). After specifying the taxa of interest, QUIDDICH’s functions 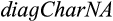 and
and 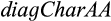 can be used to extract four different types of diagnostic genetic characters. Assuming that
can be used to extract four different types of diagnostic genetic characters. Assuming that 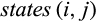 and
and 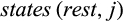 denote the sets of all states that are present in the j-th column of the alignment in any row that belongs to taxon i or the remaining taxa, respectively, these types are defined as follows:
denote the sets of all states that are present in the j-th column of the alignment in any row that belongs to taxon i or the remaining taxa, respectively, these types are defined as follows:
Definition 1.Type 1 characters distinguish each individual of the taxon i of interest from all individuals of the remaining taxa and are fixed for one state in taxon i. Mathematically, the j-the column of a given labeled alignment is a type 1 character of taxon i if (a) 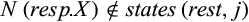 , (b)
, (b) 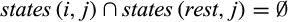 , and (c)
, and (c) 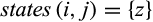 with
with 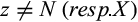 .
.
Definition 2.Type 2 characters distinguish each individual of taxon i from all individuals of the remaining taxa and are not fixed for one state in taxon i. Mathematically, the j-th column of a given labeled alignment is a type 2 character of taxon i if (a) 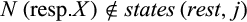 , (b)
, (b) 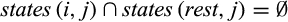 , (c)
, (c) 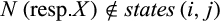 , and (d)
, and (d) 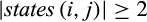 .
.
Definition 3.Type 3 characters distinguish some (but not all) individuals of taxon i from all individuals of the remaining taxa. Mathematically, the j-th column of a given labeled alignment is a type 3 character of taxon i if (a) 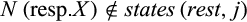 , (b)
, (b) 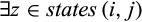 with
with 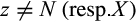 and
and 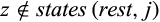 , and (c) it is not a type 1 or 2 character of taxon i.
, and (c) it is not a type 1 or 2 character of taxon i.
Definition 4.Type 4 (or pairwise diagnostic) characters distinguish each individual of taxon i from all individuals of at least one (but not all) other taxon while being fixed in both the taxon of interest and the compared taxa. Mathematically, the j-th column of a given labeled alignment is a type 4 (or pairwise diagnostic) character of taxon i if (a) 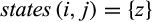 with
with 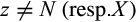 , (b) there is a taxon l with
, (b) there is a taxon l with 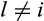 , such that
, such that 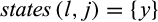 with
with 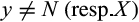 and
and 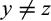 , and (c) it is not a type 1, 2, or 3 character of taxon i.
, and (c) it is not a type 1, 2, or 3 character of taxon i.
It is to note that QUIDDICH’s type 1 and type 2 characters are similar to CAOS’s homogeneous and heterogeneous simple pure characteristic attributes (CAs), while type 3 characters are similar to simple private CAs. SPIDER combines the first two types as pure, simple diagnostic nucleotides, but does not consider type 3. Apart from this, neither CAOS nor SPIDER considers type 4 characters.
The reasoning behind the definitions is as follows: If an arbitrary taxon 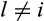 is masked at position j, it is impossible to know for sure that taxon i and taxon l do not share any character states at this position as
is masked at position j, it is impossible to know for sure that taxon i and taxon l do not share any character states at this position as 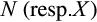 denotes an unknown state that may be replaced by any other symbol. Hence, the first condition in Definitions 1 to 3 and the second condition
denotes an unknown state that may be replaced by any other symbol. Hence, the first condition in Definitions 1 to 3 and the second condition 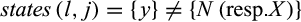 from Definition 4 are necessary to ensure the distinctness of taxon i and the taxa it is compared to. In addition to this, diagnostic characters must fulfill
from Definition 4 are necessary to ensure the distinctness of taxon i and the taxa it is compared to. In addition to this, diagnostic characters must fulfill 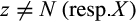 for at least one state
for at least one state 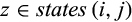 , see Condition (c) of Definitions 1 and 2, Condition (b) of Definition 3, and Condition (a) of Definition 4. This is necessary because a state that is unknown cannot be characteristic for taxon i. The last condition in each definition ensures that a character cannot be of more than one type.
, see Condition (c) of Definitions 1 and 2, Condition (b) of Definition 3, and Condition (a) of Definition 4. This is necessary because a state that is unknown cannot be characteristic for taxon i. The last condition in each definition ensures that a character cannot be of more than one type.
The definition of type 4 characters extends the suggestion of Zielske and Haase (2015) by adding Condition c) and considering indels. It is also to note that type 4 characters are not “symmetric,” that is, if the j-th column is found to be a type 4 character of taxon i when being compared to taxon l, it is not necessarily a type 4 character of taxon l when being compared to taxon i.
The output of the functions is for each taxon 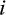 of interest a set of tuples
of interest a set of tuples  , each one representing one identified diagnostic character with j denoting its alignment position, t its type, Z the set of states that are characteristic for taxon i, that is,
, each one representing one identified diagnostic character with j denoting its alignment position, t its type, Z the set of states that are characteristic for taxon i, that is, 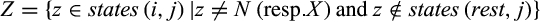 in case of type 1, 2, and 3 characters and
in case of type 1, 2, and 3 characters and 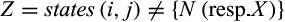 in case of type 4 characters, and Y being the set of taxa that fulfill Condition (b) of Definition 4 (only relevant for type 4 characters). The algorithm on which the functions are based and its proof of correctness can be found in the Appendix S1. If the user chooses that gaps shall not be considered as “valid” character states, that is, they cannot be characteristic for a taxon of interest, the calculations and the output are adjusted accordingly. In addition to this, QUIDDICH’s function
in case of type 4 characters, and Y being the set of taxa that fulfill Condition (b) of Definition 4 (only relevant for type 4 characters). The algorithm on which the functions are based and its proof of correctness can be found in the Appendix S1. If the user chooses that gaps shall not be considered as “valid” character states, that is, they cannot be characteristic for a taxon of interest, the calculations and the output are adjusted accordingly. In addition to this, QUIDDICH’s function 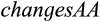 can be used to identify those diagnostic characters in a nucleotide alignment of protein-coding loci that also cause diagnostic characters in the corresponding amino acid alignment.
can be used to identify those diagnostic characters in a nucleotide alignment of protein-coding loci that also cause diagnostic characters in the corresponding amino acid alignment.
3 PERFORMANCE
Not only does QUIDDICH overcome CAOS’s and SPIDER’s drawbacks regarding the identification of diagnostic characters outlined above, it is also faster. Assume that a labeled alignment is given with r and c being the number of its rows and columns. Additionally, assume that the dataset contains 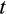 taxa, of which
taxa, of which 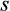 are set as taxa of interest. To extract type 1, 2, or 3 characters, the functions
are set as taxa of interest. To extract type 1, 2, or 3 characters, the functions 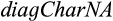 and
and 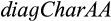 of the QUIDDICH package have an overall runtime in
of the QUIDDICH package have an overall runtime in 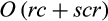 , because they first scan the alignment for polymorphic sites, which can be done in O(rc), before extracting for each combination of taxon of interest and polymorphic site the two sets
, because they first scan the alignment for polymorphic sites, which can be done in O(rc), before extracting for each combination of taxon of interest and polymorphic site the two sets 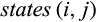 and
and 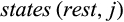 , which can be done in O(scr).
, which can be done in O(scr).
The runtime of SPIDER, which is in O(rc+tcr), can be calculated similarly. The difference is that SPIDER considers all taxa in the dataset one after the other, while  restricts the calculation to the taxa of interest.
restricts the calculation to the taxa of interest.
The runtime of CAOS is in 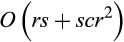 not including the manual adjustments to the tree that have to be made beforehand. The algorithm starts by numbering the nodes of the tree and conducting a Fitch optimization (Williams & Fitch, 1990) on it, both of which can be done in O(r). Then, it proceeds from the root toward the leaves calculating for each inner node n with the children n1 and n2 and each alignment column j the sets
not including the manual adjustments to the tree that have to be made beforehand. The algorithm starts by numbering the nodes of the tree and conducting a Fitch optimization (Williams & Fitch, 1990) on it, both of which can be done in O(r). Then, it proceeds from the root toward the leaves calculating for each inner node n with the children n1 and n2 and each alignment column j the sets 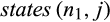 and
and 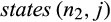 . This has to be repeated for each of the (r−1) inner nodes of the tree and each alignment column, leading to a calculation time in O(rcr). In total, we have a runtime in
. This has to be repeated for each of the (r−1) inner nodes of the tree and each alignment column, leading to a calculation time in O(rcr). In total, we have a runtime in 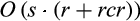 , which can be rewritten to
, which can be rewritten to 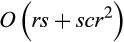 .
.
4 Application OF QUIDDICH
To examine the practicality of QUIDDICH, we investigated three datasets. The first one was an alignment of cytochrome c oxidase I (COI) of nine Pontohedyle Golikov & Starobogatov, 1972, species, interstitial marine slugs, analyzed by Jörger and Schrödl (2013) using CAOS. It can be retrieved as electronic supplementary material of their paper. Applying the function 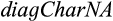 searching for all type 1 and 2 characters delivered the same results as in the paper.
searching for all type 1 and 2 characters delivered the same results as in the paper.
The second dataset (http://purl.org/phylo/treebase/phylows/study/TB2:S15532/) was an alignment of COI, 16S rRNA gene sequences, and internal transcribed spacer 2 (ITS2) of small, inconspicuous New Caledonian freshwater gastropods of the family Tateidae that was analyzed by Zielske and Haase (2015) in order to complement diagnoses of morphologically practically indistinguishable genera, again using CAOS. Searching for all type 1 and 4 characters and setting the parameter 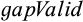 to false delivered the same diagnostic positions as in the paper.
to false delivered the same diagnostic positions as in the paper.
The third dataset (Appendix S1) was an alignment of COI comprising twelve tateid genera from New Zealand. A foregoing analysis of Haase (2008) found that Meridiopyrgus Haase, 2008, and Rakiurapyrgus Haase, 2008, as well as Hadopyrgus Haase, 2008, and Opacuincola Ponder, 1966, are almost identical regarding morphological features, while being phylogenetically very distinct. Thus, it was indicated to complement the morphological descriptions with a set of diagnostic genetic characters. The numbers of all type 1, 2, and 3 characters delivered is given in Table 1.
| Genus | Type 1 | Type 2 | Type 3 |
|---|---|---|---|
| Meridiopyrgus | 0 | 0 | 9 |
| Rakiurapyrgus | 6 | 0 | 0 |
| Hadopyrgus | 4 | 0 | 0 |
| Opacuincola | 0 | 0 | 17 |
Additionally, QUIDDICH identified 26 type 4 characters that distinguish Hadopyrgus from Opacuincola, 28 type 4 characters that distinguish Opacuincola from Hadopyrgus, 37 type 4 characters that distinguish Meridiopyrgus from Rakiurapyrgus, and 33 type 4 characters that distinguish Rakiurapyrgus from Meridiopyrgus. The comprehensive output is given as (Tables S1–S3).
5 OBTAINING QUIDDICH
QUIDDICH is a package of the statistical programming environment R (R Core Team, 2013), which can be downloaded from the comprehensive R archive network (CRAN, https://cran.r-project.org/package=quiddich) for all computing platforms. The package can also be obtained by entering the following commands into R’s console:
-
>install.packages(“quiddich”)
-
>library(quiddich)
It depends on the package “APE,” which provides the necessary data structures and basic functions. If APE is not already installed on the system, it is automatically installed when the above commands are run. The download of QUIDDICH includes a manual.
ACKNOWLEDGEMENTS
We would like to thank Mareike Fischer for her input on this contribution and two reviewers for their helpful comments.

