

Tumor Selectivity at Short Times Following Systemic Administration of a Liposomal Temoporfin Formulation in a Murine Tumor Model
ABSTRACT
Meso-tetra(hydroxyphenyl)chlorin (mTHPC) (INN: Temoporfin) is one of the most potent photodynamically active substances in clinical use. Treatment protocols for Temoporfin-mediated photodynamic therapy often rely on drug-light intervals of several days in order for the photosensitizer to accumulate within the target tissue, though tumor selectivity is limited. Here, the mTHPC localization was studied at 2–8 h following systemic administration of a liposomal Temoporfin formulation (0.15 mg kg−1 b.w.) in HT29 human colon adenocarcinoma in NMRI nu/nu mice. Photosensitizer distribution within tumor and internal organs was investigated by means of high performance liquid chromatography following chemical extraction, as well as in situ fluorescence imaging and point-monitoring fluorescence spectroscopy. For tumor tissue, the Temoporfin concentrations at 4 h (0.16 ± 0.024 ng mg−1) and 8 h (0.18 ± 0.064 ng mg−1) were significantly higher than at 2 h (0.08 ± 0.026 ng mg−1). The average tumor-to-muscle and the tumor-to-skin selectivity were 6.6 and 2, respectively, and did not vary significantly with time after photosensitizer injection. In plasma, the Temoporfin concentration was low (0.07 ± 0.07 ng mg−1) and showed no significant variation with time. Our results indicate a rapid biodistribution and clearance from the bloodstream. Within the same type of organ, data from both fluorescence methods generally exhibited a significant correlation with the extraction results.
Introduction
Photodynamic therapy (PDT) is an investigational clinical method for local treatment of certain types of malignancies in various organs. The modality exhibits interesting advantages such as the possibility of repeated treatment and restriction of the treatment-induced tissue damage to irradiated sites. In PDT, a photosensitizer is excited by absorption of light at an appropriate wavelength followed by energy transfer to oxygen molecules or other tissue constituents. The toxic radicals thus formed, of which singlet oxygen is believed to be the most important, cause tissue destruction by means of direct cell kill, vascular damage and activation of the anti-tumor immune system components (1).
Meso-tetra(hydroxyphenyl)chlorin (mTHPC) (INN: Temoporfin) is one of the most potent photosensitizers for PDT in present use (2) and it has been successfully applied in the treatment of various indications, for example the head and neck (3), prostate (4) and pancreatic (5) cancer. Strong response to Temoporfin-mediated PDT has been observed for short drug-light intervals, when the treatment outcome is mostly influenced by vascular response as the photosensitizer is located within blood vessels, vascular endothelial cells and tissue close to blood vessels (6–8). On the other hand, longer drug-light intervals, i.e. exceeding 2 days, allow the photosensitizer to diffuse further away from blood vessels and into tumor cells (8) possibly leading to a selective uptake in tumors (9,10). Clinical Temoporfin PDT therefore often relies on drug-light intervals in the order of several days during which patients are restricted to limited light exposures (3–5). This prolonged skin photosensitivity is one of the disadvantages when using Temoporfin. Due to the high affinity of the drug to bind to lipid structures, which is of importance for its relatively high PDT efficiency, the drug diffuses rapidly from the bloodstream into surrounding tissues, an effect that has been observed to cause problems at the injection site (5,7). Furthermore, the hydrophobic character of the molecule leads to poor water solubility and formation of aggregates within aqueous media (11), altering the spectroscopic and photosensitizing characteristics of the compound (12).
In order to prevent formation of aggregates, prolong photosensitizer circulating lifetimes and to improve water solubility, tumor selectivity and PDT response, several research groups have tried different delivery vehicles such as liposomes, nano-particles and conjugation to antibodies (13,14). The potential importance of avoiding aggregation was observed within a cell culture medium, where encapsulation of the hydrophobic photosensitizer bacteriochlorin a in liposomes, thereby promoting monomerization, was shown to increase oxygen consumption during PDT and decrease cell survival as compared with its partly aggregated original formulation (15). Possible evidence of mTHPC aggregation in vivo has been observed by Hopkinson et al. (16). Richter studied the use of liposomes as carrier of the hydrophobic benzoporphyrin derivative monoacid ring A (BPD-MA), reporting on higher absolute photosensitizer concentration within tumor and more pronounced PDT effect as compared with the original formulation (17). These findings were attributed to a different micro-localization within the cells and an increased association with low density lipoproteins (LDLs) when incorporated into liposomes. Pegaz et al. have recently compared two different liposomal Temoporfin formulations (Foslip, containing conventional liposomes based on dipalmitoylphosphatidylcholine [DPPC], and Fospeg, based on poly(ethylene glycol) [PEG]-modified liposomes) in terms of plasma circulation half-life times and PDT-induced photothrombic activity (18). The Temoporfin levels within the vasculature were studied up to 1200 s after photosensitizer injection, which is relevant for the treatment of age-related macular degeneration (AMD) by means of PDT. For the two formulations investigated, similar fluorescence pharmacokinetic profiles were observed, whereas the Temoporfin within the PEGylated liposome carrier proved more efficient for vascularly targeted PDT. In addition, the observation of plasma half-lives exceeding that of Visudyne®, a photosensitizer commonly used in the treatment of AMD, was ascribed to the more stable composition of the liposomes incorporating the mTHPC molecules. Fospeg has also been compared with an ethanol formulation of Temoporfin in PDT of feline skin tumors (19). In that study, the use of Fospeg demonstrated PDT response in all subjects, higher bioavailability, faster distribution and a slightly improved tumor selective uptake as compared with an ethanol formulation of Temoporfin.
An important issue in improving the understanding of various transportation vehicles for PDT photosensitizers is to be able to measure noninvasively the photosensitizer concentration. Optical methods are promising tools for tissue diagnostics and measurement of photosensitizer tissue levels in clinical environments. Fluorescence spectroscopy can provide signals related to the photosensitizer concentration and has the advantage of being a noninvasive technique that reveals results in real time (20). Fluorescence can either be studied in a point-monitoring mode (21), often using an optical fiber or a thin fiber probe for light delivery and collection, or in an imaging mode (22), where larger areas can be examined in a noncontact configuration. An imaging approach provides additional information on heterogeneities that are difficult to obtain with a point-monitoring detection technique. As Temoporfin is characterized by a relatively strong fluorescence yield, where excitation within the near-ultraviolet wavelength region results in distinctive fluorescence around 652 nm, fluorescence spectroscopy is a valuable tool in estimating photosensitizer concentration in situ within various tissue types.
In this work we present ex vivo pharmacokinetic studies following systemic administration of Temoporfin incorporated into conventional liposomes based on DPPC. The use of these liposomes provides a good biocompatibility. High performance liquid chromatography (HPLC), in this work considered as the ‘‘gold standard,’’ and fluorescence spectroscopy, both in point-monitoring and imaging mode, are used to assess Temoporfin levels in a subcutaneously implanted HT29 human colon adenocarcinoma model as well as in internal organs in a murine model. By employing these three methods, issues such as possible selectivity at short times following drug administration (2–8 h) and heterogeneity of the photosensitizer accumulation are addressed. This study adds to the previous work by Pegaz et al. (18) the use of longer time intervals after administration and to that by Buchholz et al. (19) the monitoring of Temoporfin concentration within internal organs. Furthermore, the possibilities of using noninvasive fluorescence spectroscopy to quantify Temoporfin concentration in exposed tissues are explored. The resulting photosensitizer concentrations are tested for correlation among the three methods employed and we speculate on how to improve the accuracy in absolute fluorescence level as a photosensitizer concentration estimate.
Materials and methods
Photosensitizer. Temoporfin is a dark purple, nonhygroscopic, nonsolvated crystalline powder, which is soluble in alcohol/acetone/ethyl acetate and practically insoluble in all aqueous media. The single component is of 98% purity with a molecular weight of 680.24 and a fluorescence emission peak at 652 nm. In the novel formulation used, Foslip (Biolitec Pharma Ltd, Dublin, Ireland), the hydrophobic mTHPC is bound to the membrane compartment of a phospholipid bilayer. The liposome formulation is based on DPPC, monosaccharide, water and polyoxyethylene polyoxypropylene block copolymers (18). The liposomes were reconstituted and dissolved in 3 mL of sterile water to give a photosensitizer concentration of 1.5 mg mL−1. Further dilution in 5% aqueous glucose solution provided a photosensitizer concentration of 0.075 mg mL−1 Foslip. All compounds were stored at 4°C in the dark.
Animal model. The study was performed on adult female athymic NMRI nu/nu mice (Harlan Winkelmann GmbH, Borchen, Germany). All animal experiments were carried out in compliance with the German Animal Protection Act. Six- to eight-week-old mice, weighing 22–24 g, were inoculated subcutaneously in the left and right hind thigh with a suspension of HT29 human colorectal carcinoma cells (0.1 mL of 8 × 107 cells mL−1 in 5% glucose). Experiments were performed 11 days later, when the tumors had reached a surface diameter of 5–8 mm and a thickness of 2–4 mm. Mice were injected with 50 μL of Foslip, corresponding to 0.15 mg Temoporfin kg−1 b.w., into the lateral tail vein. A dose of 50 mg kg−1 b.w. sodium pentobarbital injected i.p. was used for anesthesia. Animals were sacrificed at four different time intervals after injection of Foslip (2, 4, 6 or 8 h) and samples of plasma, spleen, liver, lung, heart, kidney, skin, muscle and the two tumors were excised for HPLC analysis. Furthermore, spleen, liver, skin, muscle and tumors were also investigated by imaging and point-monitoring fluorescence spectroscopy immediately after animal sacrifice. Both muscle and skin tissues studied were excised from regions just at the tumor periphery. All excised tissue samples were stored in darkness at −80°C until the HPLC analysis. Three animals without Foslip injection were used as controls. The number of samples investigated for each organ, time following photosensitizer administration and method of photosensitizer quantification are listed in Table 1.
| Organ/fluid | HPLC | Fluorescence point | Fluorescence imaging |
|---|---|---|---|
| Plasma | [6 3 3 3] | – | – |
| Spleen | [5* 3 3 3] | [5 3 3 3] | [6 3 3 3] |
| Liver | [6 3 3 3] | [5 3 3 3] | [6 3 3 3] |
| Lung | [6 3 3 3] | – | – |
| Heart | [6 3 3 3] | – | – |
| Kidney | [6 3 3 3] | – | – |
| Skin | [6 3 3 3] | [5 3 3 3] | [6 3 3 3] |
| Tumor | [11 6 6 3] | [11 6 5* 6] | [6 3 3 3] |
| Muscle | [6 3 3 2*] | [5 3 3 3] | [6 3 3 3] |
- *One sample identified as outlier.
HPLC analysis. All tissue samples were minced by cutting with a scalpel, weighed and freeze dried (freeze drying system Alpha 1-4 LSC; Martin Christ Gefriertrocknungsanlagen GmbH, Osterode, Germany). The resulting powdered tissue was weighed and 10–20 mg was transferred to a 2.0 mL reaction tube after which 1.5 mL of methanol:DMSO (3:5, vol:vol) was added. The samples were immediately mixed for 3 to 5 s using a vortex mixer operating at 451.58 g and then incubated at 60°C under continuous shaking for at least 12 h. All samples were spun at 16 000 g in a centrifuge for 5 min. One milliliter of the supernatant was transferred to an HPLC vial for subsequent HPLC analysis. The HPLC device had the following specifications—Pump: “System Gold, 126 Solvent Module” (Beckman Coulter, Inc., Fullerton, CA), Autosampler: “Triathlon,” Diode Array Detector: “System Gold, Module 168” (Beckman Coulter, Inc.) and a Fluorescence detector: “RF-10A XL” with interface SS420x (Shimadzu Europa GmbH, Duisburg, Germany). The fluorescence was excited at 410 nm and detected at 653 nm. The separation was carried out on a “LiChroCART 250-4” column (Merck KGaA, Darmstadt, Germany) with Purospher STAR RP-18 endcapped; 5 μm Guard column: “LiChroCART 4-4” with Purospher STAR RP-18e; 5 μm (Merck KGaA). Temperature: 30°C. The mobile phase consisted of acetonitrile:H2O + 0.1% trifluoroacetic acid = 57.5%:42.5% with a flow rate of 1 mL min−1. The tissue concentration of Temoporfin, given in ng mg−1 wet weight, was calculated from a calibration curve constructed by plotting the peak height values of Temoporfin standard solutions versus their concentrations.
Fluorescence imaging system. Fluorescence ex vivo measurements using both imaging and point detection were performed on the two tumors, skin, muscle, liver and spleen. For the imaging setup, the light source for fluorescence induction consisted of 12 light emitting CW diodes with peak emission at 405 nm. The beam radius of the spot focused onto the organs was 2.5 cm and the irradiance was approximately 30 μW cm−2. Fluorescence emitted by the tissue was recorded by a detection unit consisting of a CCD camera (C4742-80-12AG; Hamamatsu Photonics, Hamamatsu, Japan), a liquid crystal tunable filter having a full-width-half-maximum of 20 nm (LCTF VIS 20-35; Varispec, CRI, Inc., Woburn, CA) and a zoom objective lens (50 mm focal length and f/1.8; Nikon, Tokyo, Japan). The object distance was 23 cm and the field of view of the detection system was 3.2 × 4.2 cm. Room light images at 550 nm and fluorescence images at 500 and 653 nm, corresponding to wavelengths within the tissue autofluorescence and Temoporfin fluorescence bands, respectively, were collected. The exposure time was set to 3 s for each wavelength. All fluorescence images were recorded with dimmed room light to avoid any influence from background light. Background images in the absence of excitation light were also acquired using the same wavelengths and exposure times.
 (1)
(1)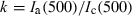 , constituting a scaling factor between data from each Temoporfin-injected animal and the mean of the three control animals.
, constituting a scaling factor between data from each Temoporfin-injected animal and the mean of the three control animals.Point-monitoring fluorescence. The Temoporfin concentration was also measured by point-monitoring fluorescence spectroscopy using an instrument described in detail in Ref. (24). Briefly, approximately 1 mW at 375 nm was delivered through a 400 μm quartz fiber with a clear-cut distal end in contact with the tissue. The induced fluorescence was collected using the same optical fiber and the reflected laser light was removed by a dichroic beamsplitter (LWP-45-RS396-TU450-700PW1012UV; CVI Technical Optics Ltd, Onchan, UK) and a long pass filter (RG395; Schott AG, Mainz, Germany). The fluorescence signal, F(λ), was detected by a spectrometer (USB4000; Ocean Optics, Dunedin, FL) and normalized at 500 nm.
 (2)
(2) (3)
(3)Statistical analysis. For each organ, the Temoporfin concentration estimate of every sample was compared with the mean value from the 15 animals. If the difference was greater than three standard deviations, the data point was considered to be an outlier and removed from the following analysis. Identified outliers are indicated in Table 1. With an ANOVA test the null hypothesis, stating that the four time points do not result in different Temoporfin concentrations, was tested for each individual organ. The agreement among the three methods used for assessing the Temoporfin concentration was quantified by studying the correlation of the HPLC data, the fluorescence contrast ratio and the Temoporfin fluorescence spectral amplitude. The hypothesis of no correlation for each organ was tested. For all tests, P < 0.01 was considered significant.
Results
HPLC results
The Temoporfin concentration as a function of time after injection is shown in Fig. 1. For all organs except the tumors, the error bars indicate the standard deviations arising due to inter-animal variations, whereas for tumor tissue, the error bars also partly reflect intra-animal differences as each animal had two inoculated tumors. For tumor tissue, the Temoporfin levels at 4 h (0.16 ± 0.024 ng mg−1) and 8 h (0.18 ± 0.064 ng mg−1) were significantly higher than at 2 h (0.08 ± 0.026 ng mg−1). Within the entire time interval, the average Temoporfin concentration was 0.12 ng mg−1, ranging between 0.04 and 0.25 ng mg−1. No difference in photosensitizer concentration was found between left and right tumors. Also, no trace of Temoporfin was found in any of the control animals. According to the ANOVA test, only within tumor tissue did the photosensitizer concentration display any significant variation with time after injection. In Table 2, the average selectivity of Temoporfin in tumor compared to other organs investigated is listed for the time points investigated. The tumor-to-muscle ratio averaged for each time point is between 5.5 and 8.1 in the time interval of 2–8 h, with the highest selectivity achieved at 2 h. The tumor-to-muscle ratio did not change significantly with time and displayed a total average of 6.6. The large variation of tumor-to-muscle selectivity at 8 h is partly due to the small number of samples. At 2 h the tumor-to-muscle ratio ranges between 2.8 and 26.5, mostly reflecting a large biological variation at this short time point. A tumor-to-skin selectivity in the order of 2 was observed, which did not vary significantly with time. The Temoporfin selectivity in tumor tissue as compared to the internal organs is low, and exhibits a slight increase for the later time points.

(a–c) Temoporfin concentration as a function of time following injection within organs investigated by HPLC. Error bars represent ±1 standard deviation (SD).
| Tumor/spleen | Tumor/liver | Tumor/lung | Tumor/heart | Tumor/kidney | Tumor/skin | Tumor/muscle | |
|---|---|---|---|---|---|---|---|
| 2 h | 0.049 ± 0.016 | 0.072 ± 0.017 | 0.19 ± 0.054 | 0.47 ± 0.10 | 0.31 ± 0.058 | 1.8 ± 0.47 | 8.1 ± 8.1 |
| 4 h | 0.092 ± 0.012 | 0.13 ± 0.016 | 0.33 ± 0.043 | 0.86 ± 0.099 | 0.56 ± 0.068 | 2.2 ± 0.45 | 6.6 ± 0.99 |
| 6 h | 0.056 ± 0.018 | 0.11 ± 0.037 | 0.24 ± 0.047 | 0.69 ± 0.13 | 0.38 ± 0.088 | 1.8 ± 0.43 | 5.5 ± 1.3 |
| 8 h | 0.11 ± 0.048 | 0.17 ± 0.036 | 0.52 ± 0.21 | 1.0 ± 0.34 | 0.55 ± 0.090 | 1.8 ± 0.28 | 6.2 ± 5.4 |
| P-value | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | 0.24 | 0.83 |
Fluorescence measurements
A typical fluorescence spectrum from the skin overlying the tumor 8 h after Foslip injection is shown in Fig. 2a. The tissue autofluorescence, the Temoporfin fluorescence and the total fit including the Fourier components are also shown. For the purposes of clarity, the autofluorescence component is displayed at 50% of its true value. The fluorescence peak at 635 nm, present only in skin samples, is believed to originate from endogenous porphyrins in the mouse skin. Figure 2b shows the corresponding fluorescence signal and the fitted fluorescence components from tumor tissue. Figure 2c,d illustrates the corresponding residuals, ɛ, calculated as the difference between measured and modeled data, and the error of the fit, calculated according to (Eq. 3).

Ex vivo (a) skin and (b) tumor fluorescence spectra, the fit components representing autofluorescence and Temoporfin fluorescence, and the total fit, which also includes the Fourier terms. The autofluorescence component is displayed at 50% of the true value and only every 30 data points are shown for purposes of clarity. (c) and (d) illustrate the residuals, ɛ, corresponding to (a) and (b), respectively. The dashed lines indicate ±2 SD of the residuals and E denotes the fitting error according to (Eq. 3). For the residuals, only every 10 data points are plotted for the purpose of clarity.
The fluorescence contrast ratio, R, as a function of time after injection is shown in Fig. 3a. For the control animals, the contrast ratio was not significantly different from zero. The heterogeneity in evaluated Temoporfin concentration within an organ was characterized by the relative standard deviation arising when averaging the fluorescence contrast function within each ROI. Data from liver and spleen resulted in relative standard deviations approximately four times higher compared with the other organs. The Temoporfin fluorescence amplitude, AmTHPC, resulting from the point-monitoring measurements, as a function of time after injection is shown in Fig. 3b. No peak at 653 nm was present in the spectra from any of the control animals. The average fitting errors, Ê, for each organ are also shown. For all tissue types, the fitting errors were smaller compared with the fluorescence signal amplitude, indicating a good fit. The influence of the Fourier terms on the total fit was typically less than 10% of the autofluorescence component and their appearance mostly reflected the heterogeneous blood distribution within the tissue. Furthermore, the magnitudes of Fourier components 11–15 were less than 5% of the maximum Fourier component, reflecting the smaller importance of the higher order terms in (Eq. 2).

(a) The fluorescence contrast ratio, R, as a function of time after injection. (b) Point-monitoring Temoporfin fluorescence amplitude, AmTHPC, as a function of time following injection. Also shown are the average fitting errors. Error bars represent ±1 standard deviation.
The results from the fluorescence methods demonstrate similar time dependence in tumor tissue as the HPLC data, with a significantly lower value of Temoporfin at 2 h compared with 4 and 8 h after injection. The order of the magnitudes of the estimated Temoporfin levels in investigated organs is overall similar for the three analysis methods. The average tumor-to-muscle ratios were 3.3 ± 0.92, 3.9 ± 0.93, 3.1 ± 1.1 and 4.0 ± 1.5 for the four time intervals investigated with the fluorescence imaging method and the corresponding average tumor-to-skin ratios were 5.4 ± 1.8, 7.5 ± 6.4, 2.9 ± 1.6 and 2.8 ± 0.36. For the point-monitoring method the average tumor-to-muscle ratios were 6.1 ± 2.9, 9.5 ± 8.9, 6.4 ± 3.6 and 8.4 ± 5.4 and average tumor-to-skin ratios were 6.3 ± 2.9, 10 ± 5.1, 4.9 ± 3.2 and 6.7 ± 2.1. For both fluorescence methods, the Temoporfin tumor selectivity displayed no time dependence. The different values of the selectivity are obtained with the fluorescence methods as compared to HPLC. This can be explained by the sensitivity of the optical methods to differences in tissue optical properties. The influence of optical properties in the fluorescence measurements are discussed in a later section.
Correlation of HPLC and fluorescence data
The possibility of using the fluorescence image contrast ratio or the point-monitoring Temoporfin fluorescence amplitude as absolute photosensitizer measures was investigated by studying the correlation between the Temoporfin quantities predicted by each method and the HPLC data, in this study considered ‘‘gold standard.’’Table 3 lists the correlation between data from the three methods for the individual organs as well as the P-value for testing the hypothesis of no significant correlation. The obtained correlation values and the magnitudes of the P-values indicate a significant agreement among the three methods for all organs with a few exceptions; poor correlation was noted between optical methods and HPLC for the spleen as well as between point-monitoring fluorescence and HPLC for the skin. Also, for all organs except the spleen and skin, the 95% confidence intervals of the predicted correlation coefficients greatly overlap, indicating no significant difference of the degree of correlation of data from different organs.
| Spleen | Liver | Skin | Tumor | Muscle | |
|---|---|---|---|---|---|
| HPLC vs fluo. point | 0.43 (P = 0.15) | 0.74 (P < 0.01) | 0.56 (P = 0.04) | 0.76 (P < 0.01) | 0.88 (P < 0.01) |
| HPLC vs fluo. imag | 0.53 (P = 0.05) | 0.85 (P < 0.01) | 0.70 (P < 0.01) | 0.87 (P < 0.01) | 0.78 (P < 0.01) |
| Fluo. imag vs fluo. point | 0.67 (P < 0.01) | 0.78 (P < 0.01) | 0.75 (P < 0.01) | 0.63 (P < 0.01) | 0.70 (P < 0.01) |
The co-variation between HPLC and fluorescence data for each of the organs investigated by HPLC and both fluorescence spectroscopy methods is illustrated in Fig. 4a–d. The correlation between HPLC and fluorescence data illustrated in Table 3 is also clearly visualized in Fig. 4a–d, while the disjoint data sets underline the necessity of multiple correlation curves to describe adequately the connection between the fluorescence contrast ratio or the fluorescence amplitude and the HPLC data for all organs combined. The varying slope of the correlation curves, shown by the solid lines in Fig. 4a–d, could be partly explained by differences in tissue optical properties. For example, the high blood content in the liver and spleen causes a higher overall light absorption and thus a comparatively smaller fluorescence signal. Other factors that influence the slope of the correlation curve, especially for the high Temoporfin concentrations encountered within the liver and spleen, include saturation and re-absorption of the Temoporfin fluorescence and the possible formation of less fluorescent mTHPC aggregates (11). The optically more transparent character of the remaining organs leads to a much steeper slope of the correlation curve in Fig. 4a–d. The co-variation between the two fluorescence methods is shown in Fig. 4e,f. In contrast to Fig. 4a,c, the slopes of the correlation curves in Fig. 4e vary less for the different organs. As the tissue volumes probed by the optical methods show a better overlap than that of HPLC and the fluorescence data are influenced by the tissue optical properties whereas the HPLC result is not, the slope should ideally be identical for all organs. However, the differences in excitation wavelength and detection geometry lead to slightly different tissue volumes probed by the two fluorescence methods, a fact that might limit the agreement between the optical data sets.

Scatter plots showing the correlation between (a) and (b) Temoporfin concentrations acquired from HPLC and the fluorescence imaging contrast ratio, R, calculated from (Eq. 1). (c) and (d) illustrate the co-variation between Temoporfin concentrations acquired from HPLC and the Temoporfin fluorescence amplitude, AmTHPC, obtained from point-monitoring fluorescence data. Correspondingly, (e) and (f) show the covariance of the fluorescence contrast ratio, R, and the Temoporfin fluorescence amplitude, AmTHPC. In all subplots, the markers represent data points from individual animals and the solid lines illustrate correlation curves.
Discussion
By incorporating hydrophobic photosensitizers into liposomes, improved selectivity and more pronounced PDT effect have been observed for PDT agents such as BPD-MA (17) and bacteriochlorin a (15) as compared to the original formulation. The compound Temoporfin, also a hydrophobic photosensitizer, is one of the most potent sensitizers (2) in present use. This photosensitizer possesses interesting photophysical properties, e.g. a strong absorption band in the red wavelength region and a high fluorescence yield, making it desirable to use optical methods in order to study photosensitizer pharmacokinetics and distribution. The ethanol formulation of Temoporfin is associated with prolonged general photosensitivity, limited tumor selective uptake and aggregation within aqueous media.
Here, we investigated photosensitizer distribution for short times following systemic administration of a liposomal Temoporfin formulation utilizing chemical extraction as well as fluorescence spectroscopy. In the animal tumor model used, athymic NMRI nu/nu mice with implanted HT29 human colon adenocarcinoma, both HPLC and optical methods demonstrated selectivity in Temoporfin accumulation between tumor and muscle and tumor and skin for time intervals of 2–8 h after drug administration.
The tumor-to-muscle and tumor-to-skin selectivity displayed no significant time dependence. The average tumor-to-muscle ratio observed in the present study indicates an early Temoporfin selectivity that is slightly higher than found following administration of mTHPC dissolved in ethanol, PEG and water. For example, Westerman et al. reported on a selectivity around 3–4 at 2–8 h after administration of mTHPC dissolved in an ethanol–PEG–water solution in nude mice with a human colon carcinoma model (LS174T) (27). For that study, the selectivity increased slightly for drug-light intervals exceeding 8 h. Further comparison of our results to those of Westerman et al. shows that the average tumor-to-skin selectivity displays slight improvement over the original formulation. Pharmacokinetic studies of liposome-encapsulated BPD-MA (M1 tumors in DBA/2 mice) (17) and SIM01 (28) have also reported on an improved tumor-to-muscle selectivity as compared to administration of the original formulation of the photosensitizer. In agreement with our results, neither of these PDT-agents showed improved tumor-to-skin selectivity when incorporated into liposomes. In contrast to the results published by Westerman et al., no selectivity has been observed between human mesothelioma xenograft and muscle in nude BALB/c mice (29) or only little selectivity within a mammary carcinoma model in C3D2/F1 mice (9) for similar short times after administration of Temoporfin in an ethanol–PEG-water solution. Our results thus indicate an early Temoporfin selectivity that is higher than observed for its original ethanol formulation.
Selective accumulation of liposome-encapsulated photosensitizers has previously been explained by the fact that the liposomes serve as donors of photosensitizer molecules to lipoproteins (13,14). As proliferating cells show an increased number of LDL receptors, the association of the photosensitizer with these proteins has been shown to promote selective accumulation and increased treatment efficiency (14,30,31). Conventional liposomes, such as DPPC used in the present study, are quickly opsonized by plasma proteins. Following this, the liposomes are taken up by phagocytosis and transported to organs with a rich mononuclear phagocyte system, such as the liver and spleen (13). This process also affects the bioavailability of the mTHPC molecules, supporting our observation of high Temoporfin concentration within the liver, spleen and lung. The association with the mononuclear phagocyte system could perhaps also contribute to the selectivity of Temoporfin in the tumor, as a higher degree of inflammatory cells, such as macrophages, phagocytes and leucocytes, are present in close proximity to the tumor periphery.
The photosensitizer concentration within tumors was significantly higher at 4 and 8 h than at 2 h. For the entire time interval, the average Temoporfin level in tumor tissue was 0.12 ng mg−1. This can be compared to an observed photosensitizer concentration below approximately 0.04 ng mg−1 in tumor tissue between 5 min and 6 h after administration of the original ethanol formulation of Temoporfin at the double photosensitizer dose (29). Our data for tumor tissue are also slightly higher than reported by Westerman et al. for similar time points after administration of mTHPC in ethanol, PEG and water (27). The Temoporfin levels detected in plasma were relatively low for all time points investigated, resulting in an average concentration of 0.07 ng mg−1. Cramers et al. reported on Temoporfin levels in plasma ranging between 0.3 and 2 ng mg−1 for similar time periods following systemic administration of mTHPC in an ethanol–PEG–water solution (29). Within muscle, skin, liver, lung, kidney and heart the Temoporfin levels were in the range previously published (9,27,29). Despite the use of conventional liposomes, which are known to accumulate within organs rich in mononuclear phagocytic cells, the photosensitizer concentration within the liver, spleen and lung in the present study was not higher than previously published.
In agreement with our results, a higher bioavailability has previously been noted for other hydrophobic PDT substances incorporated into liposomes (15,17,32). Maximum photosensitizer concentrations within tumor tissue, using photosensitizers incorporated with liposomes, have been shown to peak either at earlier (17), similar (28) or at later (32) time points after administration as compared to the original formulation of the photosensitizer. These differences might be due to the specific liposome and/or photosensitizer used as well as the tumor and animal model. The pharmacokinetic profile of liposome-encapsulated Temoporfin observed in the present study for short times after injection might mostly reflect the fate of the liposomes. Opsonization and association with lipoproteins or phagocytosis of conventional liposomes, such as used in the present study, are rapid processes that lead to a fast transfer of the sensitizer from the vascular compartment. These effects can explain the low photosensitizer levels in plasma and the almost flat pharmacokinetic profiles within the internal organs. Further improvements to tumor selectivity and bioavailability might be possible by prolonging the circulation time of the liposomes, for example by utilizing a pegylated liposomal formulation, also referred to as a stealth liposome (19). Another reason for the delayed concentration maximum within tumor tissue when using ethanol formulations of Temoporfin as compared to liposome-encapsulated Temoporfin could be the formation of photosensitizer aggregates in blood when administering an ethanol photosensitizer formulation. These aggregates need to be disassociated before the photosensitizer molecules can bind to plasma proteins (11,16).
The present study is limited in that photosensitizer distribution and the generalized photosensitivity were only studied up to 8 h. Within a different animal group, the photosensitizer levels displayed a decrease within the internal organs at 24 h (S. Gräfe, unpublished). These data, which were not merged with the present study due to slightly different experimental procedures, also indicated that the Temoporfin concentration within the skin, muscle and tumor did in fact not increase for this later time point. More extensive studies are needed to understand the pharmacokinetics of Temoporfin at later time points following systemic administration of this novel liposomal formulation.
In this study, HPLC was considered the ‘‘gold standard’’ to which the optical methods were compared and the data correlated. As seen in Fig. 4a–d, the extraction and fluorescence data showed a relatively good agreement within individual organs but one could also see that no single correlation curve could adequately fit the HPLC and fluorescence data for all organs. One factor that dramatically influences the overall correlation between the HPLC and fluorescence results is varying optical properties of the tissue under investigation. Optically opaque tissues, such as the liver and spleen, result in comparatively lower fluorescence signals than in for example the muscle, characterized by a higher albedo. Clearly, this indicates the importance of taking into account the influence of scattering and absorption on the detected fluorescence signal. Several authors have utilized white-light reflectance signal probing the same tissue volume as the fluorescence, to assess the tissue absorption and scattering levels. Based on this information, empirical and theoretical models have been used to solve for the intrinsic tissue fluorescence (33,34). White-light reflectance measurements could thus yield information on how to “unmask” the fluorescence amplitude and possibly improve agreement between extraction and Temoporfin fluorescence level. Another reason is that the extraction data represent the average Temoporfin concentration within the entire organ, whereas the fluorescence methods probe only the most superficial tissue regions. Furthermore, the differences in excitation wavelength and measurement geometry cause slightly different probing depths for the two optical methods. Any variation in Temoporfin concentration with depth from the exposed tissue surface in combination with varying, tissue-specific optical properties would negatively influence the agreement among the three methods both within and between individual organs. Finally, within aqueous media, the hydrophobic mTHPC molecules are known to form nonfluorescent oligomers. Despite being in monomeric form when incorporated into liposomes, it is possible that the photosensitizer molecules aggregate upon distribution within cells, especially in the case of high concentrations (35). The HPLC results are independent of such aggregation, whereas the fluorescence measurements are not, possibly resulting in the lower slope observed for the liver and spleen in Fig. 4a,c.
The liver and spleen are surrounded by a capsule rich in collagen and elastin. For the liver, its capsule was cut open and measurements were performed directly on the liver parenchyma, whereas the capsule of the spleen was kept intact during the optical measurements. The combination of differences in photosensitizer concentration between capsule and parenchymal tissue and the shallow probing depth of the fluorescence signals could explain the lack of significant agreement between HPLC and fluorescence data for the spleen. The poor correlation between point-monitoring and extraction data for skin may be explained by the difference in probing depth in combination with the layered skin structure. The measurement geometry (36) and the strong scattering within the epidermis, that mostly consists of dead cells with a low uptake of Temoporfin and high collagen content, limit the probing depth of the 375 nm light used for the point-monitoring fluorescence spectroscopy.
The two fluorescence methods showed similar agreement with the extraction data despite being based on different methods of analysis. Each method has its individual advantages. The fluorescence imaging method performs better within heterogeneous organs as it allows e.g. for spatial averaging of specific areas. The point-monitoring setup yields more detailed spectroscopic information making it possible to monitor separately individual tissue fluorophores such as tissue autofluorescence and skin porphyrin content. The ultimate system would provide full spectroscopic data in each spatial point of an image plane possibly allowing a precise delineation of tissue types. Future use of more detection bands of the LCTF would develop into such a system.
In conclusion, we have reported on significant photosensitizer selectivity between tumor and muscle tissue at 2–8 h following systemic administration of Temoporfin incorporated into conventional liposomes. The average tumor-to-muscle selectivity was slightly higher than that observed for mTHPC dissolved in ethanol, PEG and water (27) and other hydrophobic PDT agents incorporated into liposomes (17) upon investigation at similar times after injection. The tumor-to-muscle selectivity was not significantly dependent on time, whereas the ratio of photosensitizer concentration in tumor to internal organs increased for the later time points. These observations in combination with the overall low photosensitizer concentration within plasma, indicate a rapid photosensitizer distribution process. The biocompatibility of the liposomes, the rapid pharmacokinetics and the early selectivity observed for the liposome-encapsulated Temoporfin are interesting features in trying to limit the drug-light interval used clinically and need further investigation. Furthermore, we have observed that within individual homogeneous organs the Temoporfin fluorescence level, both in imaging and point-monitoring mode, can be used as a reasonable substance concentration estimate. However, when studying numerous and optically different tissues one needs to take into account the influence of background optical properties on the resulting fluorescence signal.
Acknowledgements— This work was partly funded by the EC integrated projects BRIGHT.EU FP6-IST-2003-511722 and Molecular Imaging LSHG-CT-2003-503259.

