

Ab Initio Study of Non-Peptidic Antihypertensives
Abstract
In this study, the authors report ab initio molecular orbital calculations on natural hormone angiotensin II (ANG II) that induces activity at AT1 receptor leading to vasoconstriction and subsequent hypertension. Optimized conformations and charge distributions of various conformers of natural hormone and AT1 antagonists have been studied. The major pharmacophoric features have been deduced. The charge environment of ANG II and drugs guided us in exploring the two possibilities: substrate inhibition and competitive inhibition. The results indicate that more potent drugs avoid ‘wastage’ in substrate inhibition and undergo strong competitive antagonism at the receptor. Specific binding interactions are essential for competitive antagonism. Slight differences in conformation may effect to differences in interactions with the receptor, hence modulating the antagonistic properties of the drug.
Blood pressure irregularities and associated cardiovascular problems are becoming more and more prevalent in our modern society. In recent years, a lot of work has been concentrated on designing ‘specific’ antihypertensives (1) as opposed to nonspecific drugs acting as antihypertensives (2–4). The renin–angiotensin system (RAS) is the key element in blood pressure regulation (5). Therefore, intervention of RAS at various stages (Figure 1) has been the obvious choice in designing antihypertensives (6–11). There exists overexpression of angiotensin-converting enzyme (ACE) in hypertensive patients and, consequently, angiotensin II (ANG II) is formed in more than metabolically accepted amounts to result in vasoconstriction and increased sodium ion reabsorption. The clinical and commercial success of ACE inhibitors (6,11) for the treatment of hypertension and congestive heart failure has initiated significant research in this area. Nevertheless, the side effects induced due to non-specificity of this enzyme have directed modern research in search of more specific antihypertensives (12,13). This study reports the results of ab initio molecular orbital (MO) calculations on natural substrate ANG II and AT1-specific antagonists (Figure 2). Complete conformational surfaces have been explored in each case and conformations corresponding to global minima are reported. The charge environment of natural substrate (ANG II) and non-peptidic drugs (AT1 antagonists) has been studied. Finally, investigation further explores the possibility of these drugs undergoing substrate inhibition when compared with competitive antagonism.

RAS system and targets for intervention.

AT1-specific antagonists.
Methods
Ab initio Hartree Fock MO calculations were performed on selected AT1 antagonists utilizing 6-31G* (14–19) basis set and on ANG II utilizing 3-21G (20–22) basis set. Complete geometry optimizations have been performed in each case using Berny's algorithm that utilizes redundant internal coordinates (23,24). The rms gradient required for convergence was 10−6. After complete conformational search on natural substrate and drug molecules, the global minima conformations obtained were overlapped on one another to understand key pharmacophoric features. Drug and natural substrate charge environments were studied by calculating the molecular electrostatic potential-derived charges (25). Intermolecular interaction energy [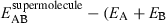 )] calculations, utilizing supermolecule approach, were performed between drug and natural substrate to explore the possibility of substrate inhibition. Several orientations of drug and natural substrate were explored. Two lowest energy conformations of natural substrate were considered for these intermolecular interaction calculations. Only the best orientation corresponding to the best interaction is reported here. Calculated interaction energies are without basis set superposition error (BSSE) estimates, as we are only interested in the relative comparison of interaction energies observed with different drug molecules. It is assumed that all the systems studied would have similar BSSE. We have recently described this methodology in detail (26).
)] calculations, utilizing supermolecule approach, were performed between drug and natural substrate to explore the possibility of substrate inhibition. Several orientations of drug and natural substrate were explored. Two lowest energy conformations of natural substrate were considered for these intermolecular interaction calculations. Only the best orientation corresponding to the best interaction is reported here. Calculated interaction energies are without basis set superposition error (BSSE) estimates, as we are only interested in the relative comparison of interaction energies observed with different drug molecules. It is assumed that all the systems studied would have similar BSSE. We have recently described this methodology in detail (26).
Results and Discussion
First, we investigated the conformational surface of ANG II which is a very flexible octapeptide. Complete potential energy surface was scanned. Only three low energy conformations were observed, which are shown in Figure 3 along with their relative energies. The twisted structure is identical to the model proposed (27) as determined relative to solution NMR data for ANG II.

Possible conformation of natural substrate ANG II.
The starting point of helical structure has been made by enforcing proper β-turns and H-bonds between 1,4 residues. It is obvious that the extended structure is higher in energy, lacking stabilization due to the secondary structure. The bioactive conformation of ANG II has not been experimentally confirmed to date. The optimized conformations of losartan, its derivatives and other non-peptidic antagonists are shown in Figure 4. To understand the basic requirements for an AT1 antagonist, we considered different conformational mappings (Figure 5). Initially, we superimposed an inactive drug over a clinically effective drug. Mapping analysis indicated that the inactivity was probably due to the inability of the ligand to interact with a hypothetical positively charged site on receptor. Next, we superimposed valsartan onto losartan. Valsartan is significantly more potent than to losartan. Mapping analysis indicated subtle conformational change for valsartan, as compared to losartan, and all critical intermolecular interactions with the receptor were correlated with key structural features of each drug. Small changes in conformation may lead to significant changes in drug interaction with the receptor. As shown in Figure 5, the superposition of all active versus inactive drugs reveals important information. The superposition of potent drugs suggests that introduction of second acidic group in a particular position is mandatory to enhance binding interactions. The superposition of inactive drugs suggests that tetrazole and second acidic group are important for drug anchoring to the receptor and binding interactions, respectively. The absence of either the tetrazole or the second acidic group results in drastic potency reduction for the drug.

Optimized conformations of various non-peptidic AT1 antagonists.

Conformational mappings.
The non-skeletal pharmacophoric features emerging from analysis of the above mappings are shown in Figure 6 and are summarized as follows (i) the length of molecule should be ∼15 Å; and (ii) the two acidic groups on the molecule should be ∼7–8 Å apart. The skeletal pharmacophoric features indicate a basic requirement of biphenyl structure with acidic substituents on each aromatic ring (and it is noted that such acid groups can be masked by ester prodrugs for improved bioavailability and efficacy in vivo).

Non-skeletal pharmacophoric features.
The electrostatic environment was studied by calculating molecular electrostatic potentials. Figure 7 shows the molecular electrostatic potentials condensed to atoms for natural substrate and some selected AT1 antagonists. However, caution must be rendered in terms of analyzing the overall charge environment of ANG II with respect to receptor interaction versus antagonist mechanisms of action. Antagonism at the receptor can take place in different ways, including: (i) the drug may bind competitively at the receptor (in an analogous fashion to ANG II) but induce antagonism, and such would imply that the drug molecule may be similar to ANG II in conformation and charge environment but yet exert key different interactions with the receptor; and (ii) the drug may also act as substrate inhibitor in terms of binding to ANG II, itself, to compromise its secondary structure, and block its binding to the receptor.

Electrostatic potential-derived charges for ANG II and some non-peptidic drugs (bright red color shows negative potential, green shows positive, and dark color shows neutral atoms).
Antagonists show negative environment near the carboxylic oxygen
This study provides calculations to explore the possibility of substrate inhibition by various drugs covering a wide range in potency from very low to moderate and high (see Table 1). The results of some potent, clinically used drugs losartan and valsartan are shown in Figure 8. Relative to conformational constraints at the receptor it is assumed that the bioactive conformation of ANG II is either the twisted or the helical (compact) form. Figure 8 depicts best the attractive interactions observed between the substrate and potent drug molecules, and their respective orientations. Only the best orientations are described in this report, although calculations were performed for several. Weak interactions of the order 2–4 kCal/mol are observed, which are not sufficient to uncoil the substrate to extended form. This indicates that the peptide substrate shows greater preference for preserving its secondary structure. Only a minute percentage of drug is likely involved in substrate inhibition or rather wasted, whereas a majority is likely undergoing competitive antagonism at the AT1 receptor. Valsartan shows the lesser probability of substrate inhibition, hence the drug is predicted to be more available for interaction at the receptor when compared with losartan. Figure 9 illustrates that tasosartan may possibly effect substrate inhibition. It is predicted to be able to block the C-terminal of ANG II to anchor with the site on receptor. Tasosartan may achieve both mechanisms of action, including efficient substrate inhibition and competitive antagonism. Losartan's p-COOH may block the C-terminal of ANG II; however, this drug lacks receptor-binding interactions and does not possess competitive antagonism. Furthermore, the Losartan inactive analog is predicted to not effect substrate inhibition either.
| Drug | IC50 (nm) | Predicted substrate inhibition (internal energy in kCal/mol) |
|---|---|---|
| Tasosartan | 1.2 | −8.18 |
| Valsartan | 2.7 | −2.71 |
| SC 5248 (Searle) | 2.8 | −2.30 |
| Losartan | 19.0 | −3.61 |
| EXP 6155 | 1600 | −5.42 |
| Losartan p-COOH | 11 000 | −10.14 |
| Losartan inactive | 28 000 | −3.70 |

Substrate inhibition by Valsartan and Losartan.

Substrate inhibition by Tasosartan and EXP6155.
The above discussion indicates that for the AT1 antagonist to be potent, the compound must possess proper binding interactions for competitive antagonism. It is predicted that a compound will be more effective if a part of it may also block C-terminal of ANG II; hence blocking the anchoring site of natural peptide substrate. However, it is understood that substrate inhibition alone cannot control the potency of the drug as hypertensive patients have overexpression of ANG II.
Conclusions
In this study, the optimized conformations and charge distributions of various conformers of natural hormone and AT1 antagonists have been studied. The major pharmacophoric features have been deduced with respect to the charge environment of ANG II and two possible mechanisms of action for AT1 antagonists, namely substrate inhibition and competitive receptor inhibition. The results indicate that more potent drugs avoid ‘wastage’ in substrate inhibition and undergo strong competitive antagonism at the receptor. Slight differences in conformation may effect to differences in interactions with the receptor; hence, modulating the antagonistic potency of the drug. Tasosartan is predicted to possibly achieve both mechanisms of action, including potent substrate inhibition and competitive antagonism.
Acknowledgements
We gratefully acknowledge financial support from DST, New Delhi, under project no. SR/S1/PC-06/2004. We also acknowledge infrastructural support by UIET, CSJM University, Kanpur.

