

In Silico Prediction of SARS Protease Inhibitors by Virtual High Throughput Screening
Abstract
A structure-based in silico virtual drug discovery procedure was assessed with severe acute respiratory syndrome coronavirus main protease serving as a case study. First, potential compounds were extracted from protein–ligand complexes selected from Protein Data Bank database based on structural similarity to the target protein. Later, the set of compounds was ranked by docking scores using a Electronic High-Throughput Screening flexible docking procedure to select the most promising molecules. The set of best performing compounds was then used for similarity search over the 1 million entries in the Ligand.Info Meta-Database. Selected molecules having close structural relationship to a 2-methyl-2,4-pentanediol may provide candidate lead compounds toward the development of novel allosteric severe acute respiratory syndrome protease inhibitors.
Severe acute respiratory syndrome coronavirus (SARS-CoV) has been recently found to be a potent pathogen of humans and capable of rapid global spread. It is a life-threatening form of pneumonia characterized by high fever, nonproductive cough, chills, myalgia, lymphopenia, and progressive infiltrates as indicated by chest radiography (1). Relative to its emergence a few years ago, an epidemic rapidly spread as facilitated by international air travel, from its origin in Guangdong Province, China, to many other countries. The World Health Organization (WHO) has reported over 8000 SARS cases and nearly 800 deaths resulting from the infection with the SARS-associated coronavirus (SARS-CoV; 2). Since about 2003, various SARS-CoV protein targets for drug discovery were identified, including SARS protease, polymerase and helicase (3). This study describes an in silico method that captures key features of potential inhibitor molecules to provide specificity and address opportunities for chemical biology and drug design (4). Our approach is based on experimental information contained in publicly available databases, therefore presenting a foundation for experimental validation. Genomic research provides an ever increasing number of potential drug targets. Structural biology allows for intense use of available and experimentally verified, structural data in various computational projects. In this study, we exploited structural homologs of SARS-CoV protease co-crystallized with small molecules to explore opportunities for drug design of potential inhibitors for this therapeutic target enzyme.
The crystal structures for all members of Structural Classification of Proteins (SCOP; 5–7), protein family of viral cysteine proteases of trypsin-fold were extracted from the Protein Data Bank (PDB) database (8,9). This family is a part of the trypsin-like serine protease superfamily that possesses closed barrel-type structure and consists of two domains of the same Greek-key duplication. The SCOP family of viral cysteine proteases encompasses three different groups of structures. The first group is 3C cysteine protease (picornain 3C) as exemplified by three proteins: human rhinovirus type 2 (1CQQ); human hepatitis A virus (1QA7, 1HAV), and Poliovirus type I (1L1N). The second group is 2A cysteine proteinase as singularly exemplified by protein Human rhinovirus 2 (2HRV). The third group is coronavirus main proteinase (3Cl-pro, putative coronavirus nsp2) as exemplified by four proteins: transmissible gastroenteritis virus (1LVO); transmissible gastroenteritis virus (1P9U); human coronavirus (1P9S); and SARS coronavirus (1Q2W, 1UJ1, 1UK2, 1UK3, 1UK4). In addition, the sequence similarity search was performed against all proteins from PDB database in order to find homologous protein structures not included in a recent version of SCOP database. The list of ligands co-crystallized with cysteine proteases presents a significant chemical diversity and includes peptides, small molecules, and inorganic salts. The ligand structures are summarized in Table 1 and includes their resolution as well as information about location of their binding inside or outside the common active site. Of these ligand structures, two peptides and one small molecule (i.e. chloroacetone) have been reported in the crystal structure of the SARS coronavirus protease (1UK4). Table 1 summarizes the known ligands co-crystalized with SCOP family of viral cysteine proteases of trypsin-fold and deposited in PDB database. Our approach considers such molecules to be relevant candidate lead compounds (or lead fragments) for further chemical modifications (e.g. peptide bond replacement by non-hydrolyzable bioisosteres). It is also noted that these initial lead compounds do not represent a comprehensive list as they are limited to relevant structures deposited in PDB database. Furthermore, some molecules which have been reported with respect to SARS drug discovery (10–17) were not included in this investigation.
| PDB codes | PDB names | Chains | Active site | Resolution | Ligand short name |
|---|---|---|---|---|---|
| 1CQQ | Human rhinovirus type 2 | A | 1.85 | ||
| + | AG7 | ||||
| 1QA7 | Human hepatitis A virus | A, B, C, D | 1.9 | ||
| + | ACE | ||||
| + | VAL | ||||
| + | NFA | ||||
| − | DMS | ||||
| − | GOL | ||||
| 1HAV | Human hepatitis A virus | A, B | 2.0 | ||
| + | OCS | ||||
| − | CL | ||||
| 1L1N | Poliovirus type I | A, B | 2.1 | ||
| 2HRV | Human rhinovirus 2 | A, B | 1.95 | ||
| − | ZN | ||||
| 1LVO | Transmissible gastroenteritis virus | A, B, C, D, E, F | 1.96 | ||
| − | DOX | ||||
| + | MPD | ||||
| − | SO4 | ||||
| 1P9U | Transmissible gastroenteritis virus | A, B, C, D, E, F | 2.37 | ||
| + | MPD | ||||
| − | SO4 | ||||
| + | CH2 | ||||
| + | Short peptide | ||||
| 1P9S | Human coronavirus | A, B | 2.54 | ||
| + | DOX | ||||
| +/− | MSE | ||||
| 1Q2W | SARS coronavirus | A, B | 1.86 | ||
| − | MPD | ||||
| 1UJ1 | SARS coronavirus | A, B | 1.9 | ||
| 1UK3 | SARS coronavirus | A, B | 2.4 | ||
| 1UK2 | SARS coronavirus | A, B | 2.2 | ||
| 1UK4 | SARS coronavirus | A, B | 2.5 | ||
| + | ATO | ||||
| GH+, K− | Short peptides |
- SCOP, Structural Classification of Protein; PDB, Protein Data Bank; SARS, severe acute respiratory syndrome.
The swiss pdbviewer software (18) was used to align analyzed structures in three-dimensional (3D) space. First, we divided all available protein chains into single domains. The domains were then structurally aligned in order to analyze the binding mode of each ligand in their active sites. From aligned structures we extracted the 1UK4 protein active site with all ligands located within it. In our drug design strategy, we used 1UK4 as the template for a flexible docking experiment in order to adjust the conformation of ligands in the new structural context. The target structure with structurally aligned ligands (co-crystallized with structurally homologous proteins) was then subjected to a further analysis in order to address any inconsistencies in the PDB database entries for certain ligands. In some instances, ligands co-crystallized in protein structures deposited in the PDB database were lacking defined atoms or functional groups may have deformed 3D representation. In the case of SARS-CoV mPro enzyme (1UK4), the ligand molecule, chloroacetone, was represented only by the chlorine atom, hence oxygen and carbon atoms needed to be added to obtain a complete molecule. In the same structure (1UK4), a pentapeptide inhibitor was contained in the crystal structure; however, its C-terminus (a carboxylic acid) was deformed to such a degree that the carbon atom had sp3 instead of sp2 hybridization. Moreover, the distance between the two oxygen atoms in the terminal carboxylate moiety was only 0.740 Å and 1.066 Å for chains G and H, respectively. The original study (19) provides information that one should expect a typical carboxylic moiety at C-terminus of the inhibitory pentapeptide, hence further analysis required prior manually remodeling to reconstitute the desired molecules.
We have explored the use of structural information contained in PDB database for an in silico virtual drug discovery campaign using, as a case study, the main protease of SARS-CoV. A similar approach using HIV protease as a case study has been reported in Refs (20,21). Two in silico methods were employed to evaluate the gathered structural information from PDB database. The first method was Electronic High-Throughput Screening (eHiTS), an exhaustive flexible docking method that systematically covers the significant part of the conformational and positional search space to produce highly accurate docking poses at a speed practical for virtual high-throughput screening (22). The second method was the Ligand.Info, a system designed for fast, sensitive, virtual high-throughput screening of small-molecule databases (23). The Ligand.Info search algorithm is based on two-dimensional structure similarity. The developed system enables search for similar compounds in a large Ligand.Info Meta-Database (24) that contains various publicly available sets of small molecules, including: (i) Harvard's ChemBank, which encompasses bioactive compounds and FDA-approved drugs (25); (ii) ChemPDB – ligands marked as Hetero Atoms in PDB files (26,27); (iii) KEGG Ligand – molecules which are found in the KEGG pathways (28); and (iv) the Open National Cancer Institute database (29). The total size of the Meta-Database exceeded 1 million entries.
Using this method, plausible inhibitors were generated as based only on the set of ligands from crystallized complexes of a protein target and other proteins from its structurally homologous family. The docking was performed on small molecules and short peptides extracted from protein–ligand complexes of the viral cysteine proteases of trypsin-fold. These small molecules and peptides were then modified in order to correct chemical attributes of the ligand structures. This set of inhibitors was next evaluated by the eHiTS flexible docking algorithm. The top-ranked ligands are summarized in Table 2 (see Figure 1 for chemical structures). The best scoring compounds of the first set were the original AG7 ligand (eHiTS docking score of −5.615) and two analogs of AG7 (eHiTS docking scores of −5.103 and −4.803, respectively). The fourth and fifth best scoring compounds were the original peptides of SARS target (PDB code: 1UK4, chains H and G) and reflected improving the initial 3D structure (eHiTS docking scores of −4.795 and −4.703, respectively). In the case of the crystal structure 1UK4, the same short peptide (chains G and H) interacting with the enzyme can be seen. In the crystal structure this peptide is present in two different conformations, and when each are used as staring points the the eHiTS results were determined to be different (cf. peptides type 1 chain H and type 1 chain G in Table 1). The same effect was observed for a very simple and flexible molecule, 2-methyl-2,4-pentanediol (MPD), which exists in crystal structures in various conformations and when used as starting points for eHiTS calculations yielded different results. Thus, eHiTS results depend on the choice of a ligand initial conformation for several reasons, including: (i) some parts of the molecule recognized by eHiTS as rigid blocks may not be rigid and should not be treated as rigid; (ii) some flexible parts may not be flexible enough; and (iii) the search may not be adequately comprehensive. We tend to think that the dependence of results on initial conformation stems from the combination of (i) and (iii) as rings within molecules being virtually identical between various poses produced by the program. Unfortunately, such a suggestion could not be confirmed as we could not identify any information relative to any parts of a molecule being treated by eHiTS as rigid blocks. Further, each molecule shown in Table 1 was used for screening the Ligand.Info Meta-Database as a query. Unfortunately, almost all designed potential lead compounds did not have any close analog in any Meta-Database subset. Experimental confirmation will obviously require chemical synthesis and biologic testing. However, MPD allosteric inhibitor was an exception as 21 similar analogs (with MTC ≥ 0.60) were found using MPD as a query.
| Ligand short name | Ligand smiles | Ligand structure | Dockingscore |
|---|---|---|---|
| AG7 | CCOC(=O)CC[C@H](C[C@@H]1CCNC1=O)NC(=O)[C@@H] (CC(=O)[C@@H](NC(=O)C2=NOC(C)=C2)C(C)C)Cc3ccc(F)cc3 |  |
−5.615 |
| AG7 version 1 | CCOC(=O)CC[C@H](C[C@@H]1CCNC1=O)NC(=O)[C@@H] (CC(=O)[C@@H](NC=O)C(C)C)Cc2ccc(F)cc2 |  |
−5.103 |
| AG7 modified version 1 | CCOC(=O)CC[C@H](C[C@@H]1CCNC1=O)NC(=O)[C@@H](C[C@H](O)[C@@H](NC=O)C(C)C)Cc2ccc(F)cc2 | 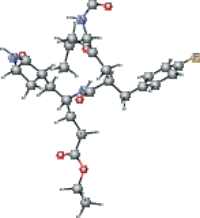 |
−4.803 |
| Peptide type 1 chain H (1UK4 protein) modified | CC(C)C[C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)[C@@H](C)O)C(=O)N[C@@H] (CCC(N)=O)C(O)=O | 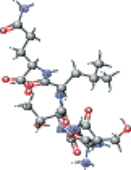 |
−4.795 |
| Peptide type 1 chain G (1UK4 protein) modified | CC(C)C[C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC (=O)[C@@H](N)CC(N)=O)[C@@H](C)O)C(=O)N[C@@H] (CCC(N)=O)C(O)=O | 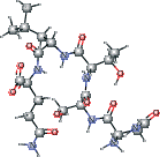 |
−4.703 |
| Peptide type 1 chain G (1UK4 protein). C-end with dioxirane ring present in the PDB | CC(C)C[C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC (=O)[C@@H](N)CC(N)=O)[C@@H](C)O)C(=O)N[C@@H] (CCC(N)=O)C1OO1 | 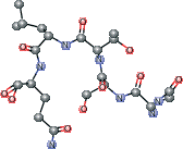 |
−4.608 |
| VNSTLQ modified version 2 | CC(C)C[C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC (=O)[C@H](CC(N)=O)NC(=O)[C@@H](N)C(C)C)[C@@H](C)O) C(=O)N[C@@H](CCC(N)=O)C(O)=O | 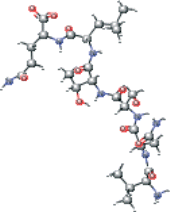 |
−4.344 |
| AG7 modified version 3 | CC[C@@H](CCC(O)=O)NC(=O)[C@H] (C)C[C@H](O)[C@@H](NC(C)=O)C(C)C | 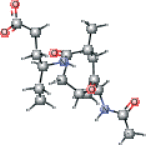 |
−4.239 |
| Peptide type 1 chain H (1UK4 protein). C-end with dioxirane ring present in the PDB | CC(C)C[C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)[C@@H](C)O)C(=O)N[C@@H] (CCC(N)=O)C1OO1 | 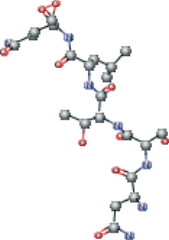 |
−3.979 |
| VNSTLQ modified version 1 | CC(C)C[C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](N)C(C)C)[C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C=O | 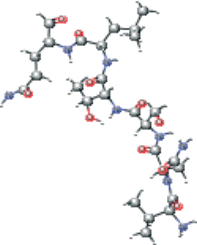 |
−3.965 |
| AG7 connected with NFA version 2 modified | CC[C@@H](CCCC(=O)N[C@@H](Cc1ccccc1)C(N)=O)NC(=O)[C@H](C)CC(=O)[C@@H](NC(C)=O)C(C)C | 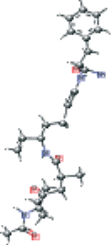 |
−3.921 |
| AG7 version 3 | CC[C@@H](CCC(O)=O)NC(=O)[C@H](C)CC(=O)[C@@H](NC(C)=O)C(C)C | 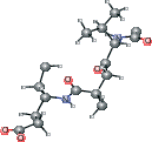 |
−3.896 |
| VNSTLQ modified | CC(C)C[C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](N)C(C)C)[C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C=O | 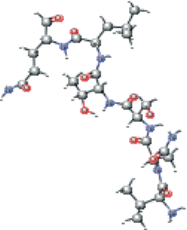 |
−3.771 |
| AG7 modified version 2 | CCOC(=O)CC[C@H](CCC(N)=O)NC(=O)[C@H](C)CC(=O)[C@@H](NC(C)=O)C(C)C | 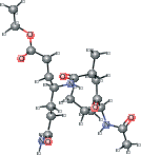 |
−3.769 |
| AG7 version 2 | CCOC(=O)CC[C@H](CCC(N)=O)NC(=O)[C@H](C)C[C@H](O)[C@@H](NC(C)=O)C(C)C | 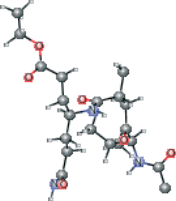 |
−3.735 |
| VAL connected with NFA modified | CC(C)[C@H](N)C(=O)N[C@@H](Cc1ccccc1)C(N)=O | 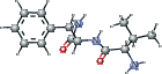 |
−3.697 |
| VAL connected with NFA | CC(C)[C@H](N)C(=O)N[C@@H](Cc1ccccc1)C(N)=O | 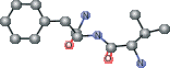 |
−3.690 |
| AG7 connected with NFA version 1 modified | CC[C@@H](CC∖C=C∖N[C@@H](Cc1ccccc1)C(N)=O)NC(=O)[C@H](C)CC(=O)[C@@H](NC(C)=O)C(C)C | 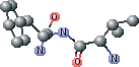 |
−3.569 |
| NFA modified | NC(=O)[C@H](Cc1ccccc1)NC=O | 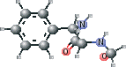 |
−3.390 |
| VAL connected with NFA ver B | CC(C)[C@H](N)C(=O)N[C@@H](Cc1ccccc1)C(N)=O | 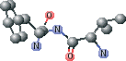 |
−3.313 |
| NFA modified version 1 | NC(=O)[C@H](Cc1ccccc1)NC=O | 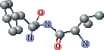 |
−3.045 |
| AG7 connected with NFA version 1 | CC[C@@H](CC∖C=C∖N[C@@H](Cc1ccccc1)C(N)=O)NC(=O)[C@H](C)C[C@H](O)[C@@H](NC(C)=O)C(C)C | 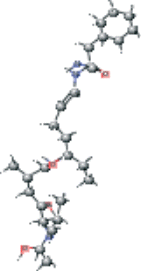 |
−2.484 |
| MPD ver 4005 | C[C@@H](O)CC(C)(C)O | 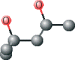 |
−2.316 |
| MPD ver 4006 | C[C@@H](O)CC(C)(C)O | 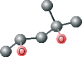 |
−2.202 |
| MPD ver 4001 | C[C@@H](O)CC(C)(C)O | 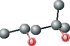 |
−2.131 |
| MPD ver 1004 | C[C@@H](O)CC(C)(C)O | 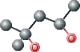 |
−2.029 |
| MPD ver 1003 | C[C@@H](O)CC(C)(C)O | 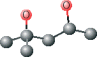 |
−2.005 |
| MPD modified | C[C@@H](O)CC(C)(C)O | 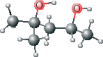 |
−1.892 |
| MPD ver 1002 | C[C@@H](O)CC(C)(C)O | 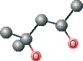 |
−1.819 |
| MPD ver 1001 | C[C@@H](O)CC(C)(C)O | 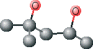 |
−1.709 |
| MPD ver 4002 | C[C@@H](O)CC(C)(C)O | 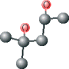 |
−1.709 |
| MPD ver 4004 | C[C@@H](O)CC(C)(C)O |  |
−1.677 |
| MPD ver 4003 | C[C@@H](O)CC(C)(C)O | 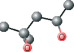 |
−1.645 |
| Chloroacetone | CC(=O)CCl | 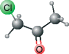 |
−1.306 |

Two dimensional chemical structures presented in Table II: AG7 (A.1), modified AG7 (A.2), NSTLQ (A.3), VNSTLQ (A.4), modified AG7 (A.5), NSTLQ modified (A.6), VNSTLQ modified (A.7), modified AG7 connected with NFA modified (A.8), other version of modified AG7 (B.9), different modification of AG7 (B.10), other modified AG7 (B.11), modified NFA connected with VAL (B.12), modified AG7 connected with NFA (B.13), modified AG7 connected with modified NFA (B.14), modified NFA (B.15), 2-methyl-2,4-pentanediol (MPD) compound (B.16), and Chloroacetone (B.17).
In conclusion, a series of lead compounds as potential SARS protease inhibitors have been preliminarily identified using a structure-based in silico virtual drug discovery approach. However, it is stressed that no MPD analogs have yet been reported to date relative to SARS protease inhibitor drug discovery (10–17,30–34). Also importantly, MPD is a chemical additive used for crystallization of biologic macromolecules, and it has been determined in co-structures with varying proteins, and not limited to only the SCOP family of viral cysteine proteases of trypsin-fold (35–43). Relative to SARS protease, MPD may provides a candidate lead compound (or fragment) for drug discovery. Several of the selected MPD analogs identified in this study are being tested experimentally (SEPSDA Sino-European Commission Project) with respect to their potential SARS protease inhibitory properties.
Acknowledgments
This work was supported by EC BioSapiens (LHSG-CT-2003-503265) and EC SEPSDA (SP22-CT-2004-003831) 6FP projects, EMBO Installation Grant to KG as well as the Polish Ministry of Education and Science (PBZ-MNiI-2/1/2005 and 2P05A00130). MvG would like to thank the Foundation for Polish Science for the fellowship.

