

Melatonin improves cardiovascular function and ameliorates renal, cardiac and cerebral damage in rats with renovascular hypertension
Abstract
Abstract: The effect of melatonin was investigated in an angiotensin II-dependent renovascular hypertension model in Wistar albino rats by placing a renal artery clip (two-kidney, one-clip; 2K1C), while sham rats did not have clip placement. Starting either on the operation day or 3 wk after the operation, the rats received melatonin (10 mg/kg/day) or vehicle for the following 6 wk. At the end of the nineth week, after blood pressure (BP) and echocardiographic recordings were obtained, plasma samples were obtained to assay lactate dehydrogenase (LDH), creatine kinase (CK), antioxidant capacity (AOC), asymmetric dimethylarginine (ADMA), and nitric oxide (NOx) levels. In the kidney, heart and brain tissues, malondialdehyde (MDA) and glutathione (GSH) levels, superoxide dismutase (SOD), catalase (CAT), myeloperoxidase (MPO) and Na+-K+ ATPase activities were determined. 2K1C caused an increase in BP and left ventricular (LV) dysfunction. In hypertensive animals LDH, CK, ADMA levels were increased in plasma with a concomitant reduction in AOC and NOx. Moreover, hypertension caused a significant decrease in tissue SOD, CAT, and Na+, K+-ATPase activities and glutathione content, while MDA levels and MPO activity were increased in all studied tissues. On the other hand, both melatonin regimens significantly reduced BP, alleviated oxidative injury and improved LV function. In conclusion, melatonin protected against renovascular hypertension-induced tissue damage and improved cardiac function presumably due to both its direct antioxidant and receptor-dependent actions, suggesting that melatonin may be of therapeutic use in preventing oxidative stress due to hypertension.
Introduction
The renin angiotensin system is crucial for the maintenance of blood pressure (BP), fluid, and sodium homeostasis and thus, plays a major role in the pathogenesis of hypertension. Renovascular hypertension, a relatively rare form of secondary hypertension, is associated with the activation of renin angiotensin system as a result of reduced renal blood flow and perfusion pressure due to renal artery stenosis [1]. The immediate hypertensive effects of angiotensin II (Ang II) occur as a result of vasoconstriction and antinatriuresis [2]. However, in the long-term Ang II causes remodeling of the arterial vasculature by vascular remodeling, accelerated atherogenesis, extracellular matrix deposition and glomerulosclerosis [3], all of which may contribute to the progression of cardiovascular and renal damage beyond the effects of high BP alone [4]. Experimental evidence suggests that reactive oxygen species (ROS), such as superoxide anion and hydrogen peroxide (H2O2), which occur as a consequence of renal artery stenosis, play a significant role in the hypertensive and hypertrophic responses to Ang II [5–8].
In physiological conditions, ROS are produced at low concentrations and function as signaling molecules to maintain vascular integrity by regulating endothelial function and vascular contraction/relaxation [9]. However, under pathological conditions, increased ROS bioactivity leads to endothelial dysfunction, increased contractility, vascular smooth muscle cell (VSMC) growth, monocyte invasion, lipid peroxidation, inflammation, and increased deposition of extracellular matrix proteins, important factors in hypertensive vascular damage [10]. It is well known that both endothelial cells and VSMC are capable of producing superoxide anions (O2–•) and other oxygen-derived radical metabolites [7]. Increased vascular O2−• production in Ang II-induced hypertension was shown to reduce the biological activity of endothelium-derived nitric oxide (NO) [11]. On the other hand, increased production of H2O2 in response to Ang II also plays a critical role in the development of vascular smooth muscle hypertrophy [6]. Structural alterations in the microcirculation, which are considered as the cause of hypertension, are also accepted as the basis of hypertension-related target organ damages in the brain, heart, or kidneys [12].
Melatonin, the major product of the vertebral pineal gland, functions as a modulator of sleep, sexual behavior, immune function, and circadian rhythms. Following the identification of melatonin (N-acetyl-5-methoxytryptamine) by Lerner et al. [13], it has been shown that melatonin is involved in the regulation of many physiological systems including cardiovascular system [14, 15]. Melatonin reduces BP [16–19] and has an anti-adrenergic action on myocardial contractility [20], which are mediated by its receptors in the heart [21] and arteries [22]. On the other hand, acting directly as an electron donor, melatonin scavenges free radicals, stimulates antioxidant enzyme systems [23, 24] and along with its metabolites it has powerful anti-inflammatory properties proven to be highly effective in reducing oxidative stress and inflammation [25, 26].
In the light of above findings, we tested the hypothesis that melatonin could alleviate renovascular hypertension through its antioxidant effects, as well as by its modulatory role on cardiovascular functions. Therefore, in an angiotensin II-dependent renovascular hypertension model, therapeutic effects of melatonin on cardiovascular functions, as well as its protective effects against oxidative damage in target organs were evaluated using various physiological and biochemical parameters.
Materials and methods
Animals
All experimental protocols were approved by the Marmara University Animal Care and Use Committee. Male Wistar albino rats (200–250 g) were kept at a constant temperature (22 ± 1°C) with 12 hr light and dark cycles and fed a standard rat chow.
Surgery and experimental design
Two-kidney, one-clip (2K1C) has been studied as an Ang II-dependent model of renovascular hypertension with elevated circulating levels of Ang II and high Ang II concentration in the cortical tissue of the clipped and non-clipped kidneys [27]. Clipping of the left renal artery and sham surgery were performed as previously described [28]. Briefly, a silver clip (internal diameter 0.25 mm) was placed around the left renal artery (2K1C group; n = 24) of the rats that were anesthetized with ketamine (100 mg/kg) and chlorpromazine (0.75 mg/kg) given intraperitoneally (i.p.). In the sham-operated control group (n = 8), animals had similar surgical procedures without clip placement. Starting on the day of surgery, rats received either melatonin (10 mg/kg/day; i.p.) or vehicle (1 mL/kg/day saline with 1% alcohol) for 9 wk (early treatment), while in a subgroup of 2K1C rats melatonin treatment was started at the end of the third week following the surgery and continued for the remaining 6 wk (late treatment).
To obtain basal readings, systolic BP and heart rate (HR) recordings were obtained in all rats before the surgical procedures, and these measurements were repeated at the end of the third and nineth weeks after the surgeries. At the end of the nineth week, echocardiographic measurements were done in all rats before they were decapitated. Trunk blood was collected and immediately centrifuged at 3000 g for 10 min to assay the levels of lactate dehydrogenase (LDH), creatine kinase (CK), antioxidant capacity (AOC), asymmetric dimethylarginine (ADMA) and nitric oxide (NO) metabolites in the plasma. In order to evaluate target organ injury, renal, cardiac and cerebral tissue samples were taken and stored at −80°C to determining the superoxide dismutase (SOD), catalase (CAT), malondialdehyde (MDA) and glutathione (GSH) levels, along with myelopreoxidase and Na+, K+-ATPase activities.
Measurement of blood pressure
Indirect BP measurement was made by the tail cuff method (Bipoac Systems, Goleta, CA, USA) before the surgery and at the end of third and nineth weeks following surgery. Initially, the rats were placed for 10 min in a chamber heated to 35°C. Then the rats were placed in individual plastic restrainers and a cuff with a pneumatic pulse sensor was wrapped around their tails. Blood pressure recorded during each measurement period was averaged from at least three consecutive readings on that occasion obtained from each rat.
Echocardiography
Echocardiographic imaging and calculations were done according to the guidelines published by the American Society of Echocardiography [29] using a 12 MHz linear transducer and 5–8 MHz sector transducer (Vivid 3,General Electric Medical Systems Ultrasound, Tirat Carmel, Israel). Under ketamine (50 mg/kg, ip) anesthesia, measurements were made from M-mode and two-dimensional images obtained in the parasternal long and short axes at the level of the papillary muscles after observation of at least 6 cardiac cycles. Interventricular septal thickness (IVS), left ventricular diameter (LVD) and left ventricular posterior wall thickness (LVPW) were measured during systole (s) and diastole (d). Ejection fraction (EF), fractional shortening (FS) and left ventricular mass (LVM) and relative wall thickness (RWT) were calculated from the M-mode images using the following formulas: 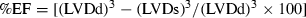 ;
; 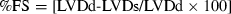 ;
; 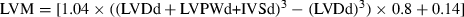 ;
; 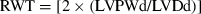 .
.
Plasma assays
Plasma levels of LDH and CK were determined spectrophotometrically using an automated analyzer (Bayer Opera Biochemical Analyzer, Leverkusen, Germany), while total AOC was measured by using a colorimetric test system (ImAnOx, Immunodiagnostic AG, Bensheim, Germany), according to the instructions provided by the manufacturer. NO metabolites (nitrates and nitrites) were assayed in plasma by the colorimetric method of Griess after enzymatic conversion of nitrates to nitrites by nitrate reductase using a colorimetric assay kit (Cayman Chemical, AnnArbor, MI, USA). ADMA concentration in plasma was measured with a highly sensitive ELISA kit (Immunodiagnostic AG). The intensity of the color reaction, measured by reading the optical density at 450 nm with a microtiter plate reader, is known to be inversely proportional with the amount of ADMA in the sample.
Measurement of tissue superoxide dismutase and catalase activities
Superoxide dismutase activity in the heart, kidney and brain samples was measured according to the previously described method [30]. Briefly, measurements were performed in cuvettes containing 2.8 mL 50 mm potassium phosphate (pH = 7.8) with 0.1 mm EDTA, 0.1 mm 0.39 mm riboflavin in 10 mm potasium phosphate (pH 7.5), 0.1 mL of 6 mm O-dianisidin.2 HCl in deionized water, and tissue extract (50, 100 μL). Cuvettes with all their components were illuminated with 20-W Slylvania Grow Lux fluorecent tubes that were placed 5 cm above and to one side of cuvettes maintaining a temperature of 37°C. Absorbance were measured at 460 nm with Schimadzu UV-02 model spectrophotometer. A standard curve was prepared routinely with bovine SOD (Sigma Chemical Co, Catalog Number: S-2515, 3000 Unit). Absorbance readings were taken at 0 and 8 min of illumination and the net absorbance were calculated.
The method for the measurement of CAT activity is based on the catalytic activity of the enzyme which catalyses the decomposition reaction of H2O2 to give H2O and O2 [31]. Briefly, the absorbances of the tissue samples containing 0.4 mL homogenate and 0.2 mL H2O2 were read at 240 nm and 20°C against a blank containing 0.2 mL phosphate buffer and 0.4 mL homogenate for about 1 min.
Measurement of tissue malondialdehyde and glutathione levels
Heart, kidney and brain samples were homogenized with ice-cold 150 mm KCl for the determination of MDA and GSH levels. The MDA levels were assayed for products of lipid peroxidation by monitoring thiobarbituric acid reactive substance formation as described previously [32]. Lipid peroxidation was expressed in terms of MDA equivalents using an extinction coefficient of 1.56 × 105/m/cm and results are expressed as nmol MDA/g tissue. GSH measurements were performed using a modification of the Ellman procedure [33]. Briefly, after centrifugation at 1500 g for 10 min, 0.5 mL of supernatant was added to 2 mL of 0.3 mol/L Na2HPO4.2H2O solution. A 0.2 mL solution of dithiobisnitrobenzoate (0.4 mg/mL 1% sodium citrate) was added and the absorbance at 412 nm was measured immediately after mixing. GSH levels were calculated using an extinction coefficient of 1.36 × 104/m/cm. Results are expressed in μmol GSH/g tissue.
Measurement of tissue myeloperoxidase activity
Since the activity of myeloperoxidase (MPO), an enzyme that is found predominantly in the azurophilic granules of polymorphonuclear leukocytes (PMN), correlates with the histochemically determined PMN in tissues, MPO activity is frequently utilized to estimate PMN accumulation in the inflamed tissues. MPO activity was measured in tissues in a procedure similar to that documented by Hillegass et al. [34]. Tissue samples were homogenized in 50 mm potassium phosphate buffer (PB, pH 6.0), and centrifuged at 41,400 g (10 min); pellets were suspended in 50 mm PB containing 0.5% hexadecyltrimethylammonium bromide (HETAB). After three freeze and thaw cycles, with sonication between cycles, the samples were centrifuged at 41,400 g for 10 min. Aliquots (0.3 mL) were added to 2.3 mL of reaction mixture containing 50 mm PB, o-dianisidine, and 20 mm H2O2 solution. One unit of enzyme activity was defined as the amount of MPO present that caused a change in absorbance measured at 460 nm for 3 min. MPO activity was expressed as U/g tissue.
Measurement of Na+, K+-ATPase activity
The activity of Na+, K+-ATPase, a membrane-bound enzyme required for cellular transport, is very sensitive to free radical reactions and lipid peroxidation. Accordingly, a reduction in Na+, K+-ATPase activity indirectly indicates membrane damage. Measurement of Na+, K+-ATPase activity is based on the measurement of inorganic phosphate released by ATP hydrolysis during incubation of homogenates with an appropriate medium. The total ATPase activity was determined in the presence of 100 mm NaCl, 5 mm KCl, 6 mm MgCl2, 0.1 mm EDTA, 30 mm Tris–HCl (pH 7.4), while the Mg2+-ATPase activity was determined in the presence of 1 mm ouabain. The difference between the total and the Mg2+-ATPase activities was taken as a measure of the Na+, K+-ATPase activity [35, 36]. The reaction was initiated with the addition of the homogenate (0.1 mL) and a 5-min pre-incubation period at 37°C was allowed. Following the addition of 3 mm Na2ATP and a 10-min re-incubation period, the reaction was terminated by the addition of ice-cold 6% perchloric acid. The mixture was then centrifuged at 3500 g, and Pi in the supernatant fraction was determined by the method of Fiske and Subarrow [37]. The specific activity of the enzyme was expressed as nmol Pi/mg protein/h. The protein concentration of the supernatant was measured by the Lowry method [38].
Statistics
Statistical analysis was carried out using GraphPad Prism 3.0 (GraphPad Software, San Diego, CA, USA). Each group consisted of 8 animals. All data were expressed as mean ± S.E.M. Groups of data were compared with an analysis of variance (ANOVA) followed by Tukey’s multiple comparison tests. Values of P < 0.05 were regarded as significant.
Results
The basal blood pressures that were recorded before the surgery were not different among the groups (Fig. 1). In the vehicle-treated group with 2K1C, the BP was significantly elevated at the third- (172 ± 6.2 mmHg; P < 0.001) and ninth- (190 ± 9.3 mmHg; P < 0.001) week recordings with respect to basal values. Similarly, at the third week measurement of the 2K1C late treatment group that has not received melatonin yet, BP was elevated (169 ± 7.5 mmHg; P < 0.001) to the levels of vehicle-treated 2K1C group. However, at the nineth week measurement of this 2K1C late treatment group, the BP was reduced significantly (144 ± 6.2 mmHg; P < 0.001), but was still higher with respect to basal values (P < 0.05). In the 2K1C group with early melatonin treatment, BP was not elevated neither at the third or nineth week measurements, indicating that melatonin treatment abolished 2K1C-induced hypertension.

Blood pressure measurements in vehicle-treated and melatonin-treated 2K1C rats. ***P < 0.001 versus basal values; +P < 0.01 versus the respective third week value. For each group n = 8.
As shown in the Table 1, recorded echocardiographic parameters including LV posterior wall thickness, LV end-diastolic and end-systolic dimensions, relative wall thickness were increased, while percent fractional shortening was decreased significantly (P < 0.01–0.001) in the vehicle-treated 2K1C group. On the other hand, melatonin treatment that was started early or late following 2K1C maintained these measurements at the control values (P < 0.05–0.001).
| Vehicle-treated control group | 2K1C group | |||
|---|---|---|---|---|
| Vehicle-treated | Melatonin-treated | |||
| Early | Late | |||
| IVS (mm) | 2.0 ± 0.05 | 2.9 ± 0.10*** | 2.2 ± 0.09+++ | 2.3 ± 0.12++ |
| PW (mm) | 1.8 ± 0.04 | 2.4 ± 0.09*** | 1.7 ± 0.07+++ | 1.8 ± 0.07+++ |
| RWT | 0.6 ± 0.02 | 1.05 ± 0.06*** | 0.7 ± 0.04+++ | 0. 8 ± 0.04++ |
| LVDs (mm) | 2.4 ± 0.12 | 3.4 ± 0.13*** | 2.4 ± 0.15+++ | 2.7 ± 0.13+ |
| LVDd (mm) | 4.0 ± 0.11 | 5.3 ± 0.15*** | 4.0 ± 0.17+++ | 4.2 ± 0.17+++ |
| EF (%) | 78.2 ± 3.0 | 63.4 ± 3.7 | 74.9 ± 1.7 | 74.2 ± 2.4 |
| FS (%) | 33.3 ± 2.1 | 22.1 ± 1.6** | 32.3 ± 2.4+ | 30.1 ± 1.7+ |
- Melatonin treatment was started either immediately after surgery (early treatment) or at the end of the third week following surgery (late treatment). Each group consists of 8 animals. IVS: interventricular septal thickness (IVS); PW: left ventricular posterior wall thickness; RWT: relative wall thickness; LVDs: left ventricular diameter in systole; LVDd: left ventricular diameter in diastole; EF: ejection fraction; FS: fractional shortening. **P < 0.01, ***P < 0.001; compared with vehicle-treated control group. +P< 0.05, ++P < 0.01, +++P < 0.001; compared with vehicle-treated 2K1C group.
Plasma LDH, CK and ADMA levels showed a significant increase in the vehicle-treated 2K1C group (P < 0.01–0.001), while treatment with melatonin beginning on the day of clipping or 3 wk after, suppressed these elevations (P < 0.05–0.001, Table 2). In addition, 2K1C groups presented with significant decreases in the plasma AOC and NO metabolite levels (P < 0.001) when treated with vehicle. However, melatonin given as an early or late treatment regimen prevented the reductions in AOC and NO metabolites (P < 0.05–0.001).
| Vehicle-treated control group | 2K1C group | |||
|---|---|---|---|---|
| Vehicle-treated | Melatonin-treated | |||
| Early | Late | |||
| LDH (U/L) | 1569 ± 71.3 | 2606 ± 149*** | 1698 ± 83+++ | 1774 ± 110+++ |
| CK (U/L) | 1180 ± 71 | 2241 ± 214*** | 1520 ± 110+ | 1607 ± 149+ |
| ADMA (μmol/L) | 0.38 ± 0.04 | 0.62 ± 0.03** | 0.42 ± 0.05+ | 0.50 ± 0.05 |
| AOC (pg/mL) | 432 ± 27.3 | 269 ± 15.1*** | 456 ± 20.8+++ | 397 ± 29.3++ |
| NO metabolites (μmol/L) | 52.1 ± 4.6 | 25.3 ± 3.3*** | 47.6 ± 3.6++ | 43.4 ± 4.2+ |
- Starting on the day of surgery, rats received either melatonin (10 mg/kg/day) or vehicle for 9 wk (early), while in some 2K1C rats melatonin treatment started at the end of the third week following the surgery and continued for the remaining 6 wk (late treatment). Each group consists of 8 animals. **P < 0.01, ***P < 0.001; compared with vehicle-treated control group. +P< 0.05, ++P < 0.01, +++P < 0.001; compared with vehicle-treated 2K1C group.
Superoxide dismutase and CAT activities measured in the cardiac, renal and cerebral tissues were reduced in the vehicle-treated 2K1C group, as compared with those of the corresponding tissues from sham-operated control group (P < 0.05–0.001; 2-4). However, in the groups with early and late melatonin treatment, 2K1C-induced reductions in both SOD and CAT activities were abolished (P < 0.05–0.01). The levels of MDA, which is a major degradation product of lipid peroxidation, were significantly increased in heart, kidney and brain tissues of vehicle-treated 2K1C group with respect to sham-operated control group (P < 0.001), while melatonin treatment caused marked decreases in the MDA levels of all tissues (P < 0.001, 2-4). Accordingly, 2K1C resulted in decreased tissue levels of GSH in the vehicle-treated group, while this reduction was significantly attenuated in melatonin-treated 2K1C groups (P < 0.05–0.001; 2-4).

(A) Superoxide dismutase (SOD) and (B) catalase (CAT) activities, (C) malondialdehyde and (D) glutathione (GSH) levels, (E) myeloperoxidase (MPO) and (F) Na+, K+-ATPase activities in the cardiac tissues of vehicle- or melatonin-treated rats with 2K1C hypertension. Melatonin treatment was started either immediately after surgery (early treatment) or at the end of the third week following surgery (late treatment). Data are presented as means ± S.E.M. *P < 0.05; **P < 0.01; ***P < 0.001 versus vehicle-treated control group; +P < 0.05; ++P < 0.01; +++P < 0.001 versus vehicle-treated 2K1C group. For each group n = 8.

(A) Superoxide dismutase (SOD) and (B) catalase (CAT) activities, (C) malondialdehyde and (D) glutathione (GSH) levels, (E) myeloperoxidase (MPO) and (F) Na+, K+-ATPase activities in the renal tissues of vehicle- or melatonin-treated rats with 2K1C hypertension. Melatonin treatment was started either immediately after surgery (early treatment) or at the end of the third week following surgery (late treatment). Data are presented as means ± S.E.M. *P < 0.05; **P < 0.01; ***P < 0.001 versus vehicle-treated control group; +P < 0.05; ++P < 0.01; +++P < 0.001 versus vehicle-treated 2K1C group. For each group n = 8.

(A) Superoxide dismutase (SOD) and (B) catalase (CAT) activities, (C) malondialdehyde and (D) glutathione (GSH) levels, (E) myeloperoxidase (MPO) and (F) Na+, K+-ATPase activities in the cerebral tissues of vehicle- or melatonin-treated rats with 2K1C hypertension. Melatonin treatment was started either immediately after surgery (early treatment) or at the end of the third week following surgery (late treatment). Data are presented as means ± S.E.M. *P < 0.05; **P < 0.01; ***P < 0.001 versus vehicle-treated control group; +P < 0.05; ++P < 0.01; +++P < 0.001 versus vehicle-treated 2K1C group. For each group n = 8.
In the vehicle-treated 2K1C group, MPO activities in the cardiac, renal and cerebral tissue samples were significantly increased when compared with those of the control group (P < 0.001; 2-4), indicating increased tissue neutrophil infiltration. However, in the 2K1C rats, which had melatonin treatment started either on the day of clipping or 3 wk after clipping, these increases in MPO activity were significantly inhibited (P < 0.01–0.001), but they were still above control levels. When compared with sham-operated control group, Na+, K+-ATPase activities measured in the heart, kidney and brain samples were reduced in the vehicle-treated 2K1C group, indicating impaired transport function and membrane damage in these tissues (P < 0.001; 2-4). On the other hand, both regimens of melatonin treatment prevented these reductions in tissue Na+, K+-ATPase activities (P < 0.05–0.01).
Discussion
In the present study, increases in lipid peroxidation and MPO activity due to Ang II-induced renovascular hypertension were accompanied by a significant reduction in tissue antioxidant defense measured by GSH, SOD, CAT and Na+, K+-ATPase activities in the cardiac, renal and brain tissues, implicating the presence of oxidative tissue damage. In addition, elevated serum levels of LDH, CK and ADMA levels, as well as reduced NO and AOC demonstrate the severity of NO-dependent oxidative stress. The results also demonstrate that melatonin treatment, as observed by the reversal of alterations in all the measured parameters, alleviated the extent of hypertension-induced oxidative organ damage and improved cardiovascular functions.
Reactive oxygen species can modulate renal hemodynamics and function both directly, by leading to vasoconstriction, and indirectly, by inducing renal inflammation. Evidence obtained from humans with essential hypertension [39] and from animal models [7, 40, 41] support the hypothesis that oxidative stress and inflammation are associated with the development of hypertension and hypertension-induced damage in target organs [12]. Oxidative stress may contribute to the generation and maintenance of hypertension via the inactivation of NO [42], which antagonizes the vasoconstrictive and pro-atherosclerotic effects of Ang II in physiological conditions. On the other hand, Ang II also decreases NO bioavailability by promoting oxidative stress. O2–•, which is the main free radical involved in the pathology of different forms of hypertension, can either cause vasoconstriction or, under certain circumstances, can interact rapidly with NO to reduce its bioavailability and further increase vasoconstriction and BP [43]. In the 2K1C hypertension model, presenting with increased plasma levels of angiotensin, high BP is usually established within 3 to 4 wk after clipping [44]. In this renovascular hypertension model, the balance in the endothelium between NO, Ang II and ROS, which is necessary for maintaining the homeostasis of the vascular wall, is altered. In turn, endothelial dysfunction due to the loss of NO bioactivity increases vascular reactivity and initiates cardiovascular inflammatory changes leading to elevation of BP [45]. It has been suggested that ADMA is an endogenous regulator of arteriolar tone through the inhibition of constitutive nitric oxide synthase (cNOS), that may be used as a significant predictor of renal blood flow and resistance [7, 46]. ADMA activates renin angiotensin system and NAD(P)H oxidase leading to increased production of O2–•, which then interferes with the bioavailability of NO, resulting in diminished dilation and increased arteriolar tone [47]. Therefore, increased ADMA is known as a potential cardiovascular risk factor indicating reduced renal plasma flow and increased renovascular resistance [48, 49]. In agreement with previous observations, 2K1C procedure in the present study resulted in elevated BP along with increased plasma levels of ADMA and increased MDA levels in various tissues, indicating the presence of endothelial dysfunction associated with NOS inhibition as well as increased lipid peroxidation. Similarly, in a group of hypertensive patients, elevated levels of ADMA and increased lipid peroxidation products along with enhanced release of ROS were suggested to be contributing to the associated microvascular endothelial dysfunction and elevated BP [50]. The causal association of oxidative stress and hypertension has also been supported by the observation that antioxidant therapy ameliorates hypertension in experimental models [51, 52].
Treatment of 2K1C rats with the antioxidant melatonin in the current study improved cardiovascular dysfunction, reversed the ADMA levels and ameliorated the oxidative tissue damage. Based on the previous studies, although it is not clear whether melatonin reduces BP by directly inhibiting ADMA production or by increasing NO bioavailability, our results show that elevated ADMA levels and reduced plasma NO levels in hypertensive rats were significantly reversed by melatonin treatment. Similarly, Girouard et al. [53] have demonstrated that melatonin increased endothelium-dependent vasodilation in mesenteric artery and aorta from both normotensive and hypertensive rats and decreased BP by improving vascular NOS pathway. The antihypertensive effect of melatonin was more pronounced than the effect of the antioxidant N-acetylcysteine [54] and was found to be comparable with the effect of spironolactone [55]. However, removing the major source of endogenous melatonin by pinealectomy caused a significant increase in BP and augmented the response to vasoconstrictor agents, while melatonin treatment lowered the BP and the increased vascular reactivity [56]. Similarly, when spontaneously hypertensive rats were treated with melatonin, a gradual decrease in BP, HR and plasma renin activity was observed [57].
Echocardiographic measurement on the nineth week of the 2K1C surgery revealed the presence of left ventricular dysfunction that accompanied high BP. Among several factors that are involved in the pathogenesis of left ventricular hypertrophy, stimulation of the renin angiotensin-aldosterone and sympathetic nervous systems and endothelial dysfunction have an important impact on ventricular functions [58]. Studies in both humans [59] and animal models [60] have demonstrated that an increase in oxidative stress might be also responsible for hypertension-induced myocardial dysfunction. Accordingly, increased Ang II activity and the associated oxidative stress might be responsible for LV dysfunction observed in rats with 2K1C in the present study. On the other hand, melatonin, when given either in the early or later phases of renovascular hypertension, decreased BP and improved heart functions, possibly through its direct antioxidant and receptor-mediated actions [61, 62]. Similarly, melatonin was shown to prevent hypertrophy and oxidative damage in the hyperthyroid rat heart [63]. On the other hand, decreased melatonin levels were reported in patients with hypertension [64], heart failure [65], ischemic heart disease [66], or acute myocardial infarction [67], suggesting the cardioprotective effects of endogenous melatonin. Taken together with the aforementioned studies, improved echocardiographic results along with the amelioration of the oxidative parameters in the current study implicate that the cardioprotective mechanisms accomplished by melatonin treatment may involve both the direct antioxidant and receptor-mediated effects of melatonin.
Reactive oxygen species have been implicated in the pathogenesis of renal injury by liberation of vasoconstrictors, direct cellular damage, and inactivation of NO, which may subsequently lead to both functional and structural renal abnormalities [68]. On the other hand, blockade of the oxidative stress pathway in an experimental model of renovascular disease was shown to improve renal blood perfusion and to prevent inflammation and fibrosis in the stenotic kidney [69]. In the present study, MDA levels in the kidneys, as well as in the cardiac and cerebral tissues, were significantly increased, indicating that oxidative stress generated in the kidneys, which contributes to the generation and maintenance of hypertension, also causes oxidative damage in multiple organs remote to the kidneys. Moreover, increased activity of Na+/K+ ATP-ase, which is a membrane-bound enzyme that requires phospholipids for its activity, also indicates lipid peroxidation-induced injury in the studied tissues [70]. As it is observed in the present study, oxidative stress reduces tissue levels of GSH and the activities of superoxide dismutase and catalase, which are essential compounds for maintaining cell integrity. It is known that deficiency or depletion of GSH and other antioxidant enzymes causes damage to macromolecules or to membrane lipids when there is consistent formation of oxygen free radicals. The results demonstrate that treatment with the antioxidant melatonin reversed the alterations in tissue MDA and GSH levels, SOD, catalase and Na+/K+ ATP-ase activities. Thus, in accordance with the previous results, the current study demonstrates that the protective effects of melatonin in hypertension-induced oxidative damage may be due to an augmentation of the endogenous antioxidants and the inhibition of lipid peroxidation by maintaining a balance in oxidant-antioxidant status. It is well established in numerous studies that melatonin displays significant beneficial actions against oxidative cellular toxicity through its capability of scavenging both oxygen- and nitrogen-based reactants and by blocking transcriptional factors, which induce pro-inflammatory cytokines [71].
Since MPO plays a fundamental role in oxidant production by neutrophils and is used as an index for the neutrophil infiltration, the increased MPO activity observed in the current study clearly demonstrates that 2K1C-induced damage in the kidneys and the two target organs is neutrophil-dependent. Activated neutrophils release MPO, causing production of large amounts of HOCl, which oxidizes and damages macromolecules, including proteins, lipids, carbohydrates and nucleic acids. Increasing evidence also suggests that neutrophils release chemotactic substances, which further promote neutrophil migration to the tissue, activate neutrophils and increase the damage [72]. Previously, melatonin was shown to reverse L-NAME-induced BP elevation and protect against cardiac oxidative injury, while depressing MPO activity [19]. In parallel to these results, current data also show that neutrophil migration plays an important role in the development of oxidative injury in hypertensive rats. Recently, it was demonstrated that melatonin directly modulates the formation of MPO intermediates and their decay rates, and thereby may serve as a potent inhibitor of MPO and its downstream inflammatory pathways [73].
In an experimental renovascular hypertension model, melatonin significantly reduced BP, improved left ventricular function and alleviated oxidative injury of the kidney, heart and brain by its ability to inhibit neutrophil infiltration, to balance the oxidant-antioxidant status, and to regulate the generation of inflammatory mediators. In conclusion, the findings of the present study support the critical pathogenic contribution of increased oxidative stress in renovascular hypertension, and suggests a role for melatonin in preserving the kidney and the target organs against hypertension-induced tissue damage.

