

THE DYNAMICS OF MITOCHONDRIAL MUTATIONS CAUSING MALE INFERTILITY IN SPATIALLY STRUCTURED POPULATIONS
Abstract
Mitochondrial genomes are usually inherited maternally and therefore there is no direct selection against mutations that have deleterious effects in males only (mother’s curse). This is true in particular for mitochondrial mutations that reduce the fertility of their male carriers, as has been reported in a number of species. Using both analytical methods and computer simulations, we demonstrate that spatial population structure can induce strong selection against such male infertility mutations. This is because (1) infertile males may reduce the fecundity of the females they mate with and (2) population structure induces increased levels of inbreeding, so that the fitness of females carrying the mutation is more strongly reduced than the fitness of wild-type females. Selection against mitochondrial male infertility mutations increases with decreasing deme size and migration rates, and in particular with female migration rates. On the other hand, the migration model (e.g., island or stepping stone model) has generally only minor effects on the fate of the mitochondrial mutations.
Mitochondria, often referred to as the “power house of the cell,” are organelles responsible for the majority of ATP production in eukaryotic cells. From an evolutionary perspective, mitochondria are interesting because they are derived from free-living bacteria and carry their own genome. In animals, this genome is particularly prone to mutations (Lynch et al. 2006), and these mutations can cause mitochondrial dysfunction that may lead to a variety of diseases (Taylor and Turnbull 2005). Given the crucial role of mitochondria in sperm development and function (e.g., Mitchell et al. 1976; De Martino et al. 1979; Cardullo and Baltz 1991; Mackenna et al. 1995), it is not surprising that mutations in mitochondrial DNA can also cause male infertility. In humans, mtDNA point mutations have been identified whose presence strongly correlates with low sperm motility or number (Spiropoulos et al. 2002; Selvi Rani et al. 2006), and male infertility can be associated with a distinct mtDNA haplogroup (Ruiz-Pesini et al. 2000; but see also Pereira et al. 2005; Bandelt 2007). Similarly, a mtDNA haplotype was found to be associated with low male fertility in a population of European hares (Smith et al. 2010). Mitochondrial mutations that impact male fertility have also been reported in mice (Nakada et al. 2006), the seed beetle Callosobruchus maculatus (Dowling et al. 2007), and Drosophila melanogaster (Clancy 2008; Clancy et al. 2011). Interestingly, in the latter two species sexually antagonistic fitness effects of mitochondrial genotypes were reported, that is, fitness effects were of opposite sign in males and females (Dowling et al. 2007; Rand et al. 2001).
Unlike nuclear genes, mitochondria are exclusively transmitted from mother to offspring in most species. This means that selection only acts on mtDNA variants that result in fitness variation in females. Mitochondrial mutations that cause male infertility but are neutral in females will therefore not be selected against and can spread in a population through random genetic drift (Frank and Hurst 1996), a phenomenon sometimes referred to as “mother’s curse” (Gemmel et al. 2004). However, this argument applies only when individuals mate randomly within the population. When there is inbreeding, we can expect that females harboring the mitochondrial mutation will also suffer to some extent from this mutation because they tend to mate more often with infertile males (e.g., their brothers) than females carrying the wild-type mitochondria. This effect can produce selection against male infertility mitochondrial mutations in inbreeding populations (Unckless and Herren 2009; Wade and Brandvain 2009). Conversely, with outbreeding (inbreeding avoidance) there can be even weak positive selection for male-deleterious mutations because males carrying the mutation reduce the fitness of wild-type females more than that of mutant females (Engelstädter and Charlat 2006). Recently, Hedrick (2011) generalized these models to arbitrary levels of assortative mating and obtained results that are in agreement with the earlier findings.
All of the above theoretical studies assume a single, undivided population. By contrast, a feature of virtually all natural populations is a certain degree of geographical population structure where individuals form subpopulations connected by migration. Population structure is well known to influence the rates of inbreeding and outbreeding in local populations (Wright 1951, 1978) and the efficacy of selection. On the one hand, population structure leads to an increased rate of inbreeding, which increases the probability of expressing recessive mutations in homozygotes and thereby increases the efficacy of selection against deleterious recessive mutations (e.g., Whitlock 2002). On the other hand, one important effect of population subdivision is local random genetic drift, which will reduce the efficacy of natural selection (e.g., Cherry and Wakeley 2003; Roze and Rousset 2003; Whitlock 2003). However, this effect is not expected to change the fixation probability of a beneficial mutation with additive effects (Maruyama 1970) unless populations are very small (Roze and Rousset 2003) or population extinctions occur (Whitlock 2003). This is because the reduced efficacy of selection is outweighed by the increased total effective population size of subdivided populations. Nevertheless, the rate of fixation of such mutations is slowed compared to a single undivided population (Cherry and Wakeley 2003; Whitlock 2003). Based on these earlier findings, we expect that population subdivision will have a major effect on the evolutionary dynamics of sexually antagonistic mutations by both impeding and enhancing selection, depending on the trade-off of selection pressures in each sex.
Here, we investigate the influence of spatial structure on the spread of mitochondrial mutations that influence male fertility. Our model covers both mutations that induce infertility in males but are neutral in females, and sexually antagonistic mutations. We derive analytical results for a limiting case of Wright’s island model (assuming an infinite number of demes) and study the full model through extensive computer simulations. In addition, we study the impact of sex-specific migration rates and of nearest-neighbor dispersal (i.e., stepping-stone models). Our results show that across a wide range of scenarios, male fertility effects in concert with pronounced population structure can strongly affect the spread of mitochondrial mutations.
Methods
THE MODEL
We consider a dioecious sexual and semelparous (nonoverlapping generations) system with traditionally defined females and males. Individuals are characterized by their mitochondrial genotype, which can be in either the “mutant ” or “wild-type” state. Mitochondria are purely maternally inherited with no paternal leakage. Individuals live in a population composed of a finite number n of demes, each containing N males and N females. The life cycle is comprised of the following events:
- 1
Mating. We assume that each female mates with one male chosen randomly from the same deme. Males may therefore mate multiple times in a population.
- 2
Reproduction. Each female produces a large number of offspring. Fertility is influenced by the genotype in a sex-specific way, in that the offspring number of mutant females is altered by a factor (1+sf) relative to wild-type females, and mutant males produce (1+sm) times as many offspring as wild-type males. These two fitness effects interact multiplicatively, so that, for example, a mutant female mating with a mutant male produces a relative number of (1+sf)(1+sm) offspring. Typically, male deleterious (sm <0, sf= 0) or sexually antagonistic mutations (sm <0, sf > 0) will be considered.
- 3
Dispersal. Male and female juveniles disperse randomly with probabilities mm and mf, respectively. The probability that a female or male juvenile is sampled in its natal deme after dispersal is given by (1−mf) and (1−mm), respectively.
- 4
Competition. Juveniles compete for survival to adulthood. This competition is sex-specific and occurs within demes, and we assume that the mitochondrial genotype does not influence survival. In each deme, N juveniles of each sex reach adulthood, whereas the remainder die. This completes the life cycle.
ANALYSIS OF THE MODEL
We derived analytical solutions for a limiting case of the full model in which we assume a very large number of demes of finite size (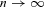 ). Analytical results for this model are derived based on methodology developed by Rousset (2004) and Reuter et al. (2008), and detailed in the Appendix.
). Analytical results for this model are derived based on methodology developed by Rousset (2004) and Reuter et al. (2008), and detailed in the Appendix.
The full model was investigated through individual-based computer simulations using the package Nemo (Guillaume and Rougemont 2006). Individual fecundity was assumed to be Poisson distributed with mean fecundity of 10. Each simulation was initialized with only wild-type individuals except for a single mutant female and a single mutant male. The simulation was then run until the mutation was extinct or had reached fixation in the population. Estimates for the fixation probability, p, were then normalized by the expected fixation probability u in panmictic populations, 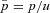 , where
, where 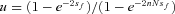 (Kimura 1962).
(Kimura 1962).
Throughout, we used a total population size of 12,000 individuals. We considered population subdivisions with deme numbers 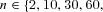 120, 240,
120, 240,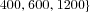 , corresponding to deme sizes
, corresponding to deme sizes 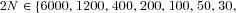
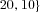 .
.
Results
ANALYTICAL RESULTS
To obtain analytical insights into our model, we studied the special case of a population with infinitely many demes of finite size. For simplicity, we assumed equal migration rates in males and females. In what follows, we give the main results of this investigation; the full derivations are detailed in the Appendix.
 (1)
(1) (2)
(2)(see eqs. A11 and A12). Here, 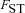 is the probability of within-deme coalescence of two individuals sampled from a deme without replacement. Equation 1 shows that the effective selection coefficient consists of two terms. The first term describes how population subdivision decreases the efficacy of selection on female fecundity through the effects of local genetic drift. Population subdivision causes increased homogeneity in allele frequencies within demes, thereby reducing this component of selection by a factor
is the probability of within-deme coalescence of two individuals sampled from a deme without replacement. Equation 1 shows that the effective selection coefficient consists of two terms. The first term describes how population subdivision decreases the efficacy of selection on female fecundity through the effects of local genetic drift. Population subdivision causes increased homogeneity in allele frequencies within demes, thereby reducing this component of selection by a factor  . By contrast, marked population subdivision—as quantified by a high value of
. By contrast, marked population subdivision—as quantified by a high value of 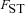 —increases the weight of the second component of selection that captures the effect of male fertility. This is because a high value of
—increases the weight of the second component of selection that captures the effect of male fertility. This is because a high value of 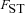 implies a high degree of inbreeding within demes, so that the fitness of mutant females will be strongly affected by the reduced fertility of the mutant males.
implies a high degree of inbreeding within demes, so that the fitness of mutant females will be strongly affected by the reduced fertility of the mutant males.
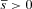 , can be expressed as
, can be expressed as
 (3)
(3)
Illustration of our analytical results on the spread of sexually antagonistic mitochondrial mutations in populations with a very large number of finite demes. In plots (A) and (B), based on equation 3, the mutation is predicted to spread in the population above the lines, and become extinct below the lines. Plot (A) shows the effect of the migration rate for a fixed deme size of N=10, whereas (B) shows the effect of deme size for a fixed migration rate of m=0.01. Plot (C) illustrates the conditions under which effective selection for a mitochondrial mutation is stronger in a structured compared to a panmictic population.
We can also ask under what conditions population subdivision enhances the spread of a mitochondrial mutation compared to the spread in a panmictic population. This will be the case when 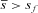 , which simplifies to smm(2−m)>sf. Figure 1C shows that for sexually antagonistic mutations, population structure always inhibits the spread of the mutation when sf>0, sm<0, but always enhances spread when sf<0, sm>0. When the sign of the selection coefficient is the same in females and males, the effect of population subdivision in enhancing or inhibiting mutation spread depends on the migration rate m, but, interestingly, not on deme size N.
, which simplifies to smm(2−m)>sf. Figure 1C shows that for sexually antagonistic mutations, population structure always inhibits the spread of the mutation when sf>0, sm<0, but always enhances spread when sf<0, sm>0. When the sign of the selection coefficient is the same in females and males, the effect of population subdivision in enhancing or inhibiting mutation spread depends on the migration rate m, but, interestingly, not on deme size N.
SIMULATION RESULTS: ISLAND MODEL WITH SEX-INDEPENDENT MIGRATION
Because the full model, assuming a finite number of demes of arbitrary size, was not tractable analytically we resorted to computer simulations. We first investigated the spread of a mitochondrial mutation that affects male fertility but is neutral in females (Fig. 2). As expected, this type of mutation behaves as a neutral mutation in an unstructured population (n=1), exhibiting a fixation probability of 1/N. However, in a structured population, the fixation probability of mutations decreasing male fertility (sm<0) is reduced and the fixation probability of mutations increasing male fertility is increased relative to that of a neutral mutation (as expected from eq. 3). This effect becomes more pronounced with decreasing deme size when the total population size is kept constant. In a highly structured population consisting of n=120 demes with 2N=100 males and females in each deme, the fixation probability is strongly altered even with moderate negative effects on male fertility.

The effect of population structure and male fitness effects on the normalized fixation probability 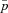 of a mitochondrial mutation that is neutral in females (sf=0). Each datapoint represents an estimate of the fixation probability that is based on
of a mitochondrial mutation that is neutral in females (sf=0). Each datapoint represents an estimate of the fixation probability that is based on 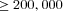 simulations. The dashed line represents a panmictic population (only a single deme) and the three solid lines represent different degrees of population subdivision (n=2, 10, and 120). The migration rate was set to m=0.1. The dotted lines represent the fixation probability derived from our analytical results (see equation 4), which was again normalized by the corresponding fixation probability in a panmictic population (i.e., 1/nN). Note that the value zero was added to the logarithmic axis to account for cases where the mutation had not become fixed in any of the simulations.
simulations. The dashed line represents a panmictic population (only a single deme) and the three solid lines represent different degrees of population subdivision (n=2, 10, and 120). The migration rate was set to m=0.1. The dotted lines represent the fixation probability derived from our analytical results (see equation 4), which was again normalized by the corresponding fixation probability in a panmictic population (i.e., 1/nN). Note that the value zero was added to the logarithmic axis to account for cases where the mutation had not become fixed in any of the simulations.
 (eq. 2) into Kimura’s (1962) formula for the fixation probability. In this case, this formula reads
(eq. 2) into Kimura’s (1962) formula for the fixation probability. In this case, this formula reads
 (4)
(4)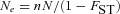 (e.g., Whitlock 2003). Interestingly, for sf=0, equation 4 simplifies to
(e.g., Whitlock 2003). Interestingly, for sf=0, equation 4 simplifies to
 (5)
(5)We next considered mitochondrial mutations with sexually antagonistic effects. Figure 3 shows the estimated relative fixation probabilities for such mutations for different deme sizes, migration rates, and male fertility effects. The spread of a mutation that moderately increases female fecundity but substantially reduces male fertility is strongly impeded when the demes are small and the migration rate not too high (Fig. 3A). When the mutation is neutral in males (Fig. 3B), the fixation probability is similar to that in a panmictic population. Finally, when the mutation is beneficial in both females and males, population structure has a positive effect on the fixation probability, which, interestingly, is strongest for intermediate migration rates (Fig. 3C). Note that in all plots in Figure 3, the case of m=1 and N=3000 (i.e., n=2) is a special case in which the mutation can never become fixed because the individuals in the two demes are merely swapped in their entirety in every generation.

Estimates of the normalized fixation probability 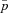 with changing deme number and migration rate for four types of mitochondrial mutations. The selection coefficient in females was fixed to sf=0.05, and the selection coefficient in males takes the values (A) sm=−0.4, (B) sm=0, and (C) sm=1. Each estimate for
with changing deme number and migration rate for four types of mitochondrial mutations. The selection coefficient in females was fixed to sf=0.05, and the selection coefficient in males takes the values (A) sm=−0.4, (B) sm=0, and (C) sm=1. Each estimate for 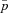 is based on
is based on 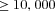 replicate simulations.
replicate simulations.
SIMULATION RESULTS: ISLAND MODEL WITH SEX-BIASED MIGRATION
In many species, females and males exhibit a different propensity to migrate. For example, migration is often male biased in mammals, whereas the opposite trend is found in birds (Greenwood 1980). It is therefore important to also consider the impact of such sex-biased migration on the spread of mitochondrial mutations.
Figure 4 shows estimates of the normalized fixation probability of different mitochondrial mutations for a wide range of female and male migration rates, mf and mm. When the mutation reduces male fertility, the fixation probability is strongly reduced unless either the male or the female migration rate is extremely high (Fig. 4A). As expected, when sm=0, the male migration rate is of no consequence for the fixation probability (Fig. 4B). Finally, when the mutation is beneficial in males, a very high male migration rate leads to a slightly reduced fixation probability, but in general, male migration rate has little effect on the fixation probability (Fig. 4C).

Estimates of the normalized fixation probability 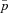 with sex-specific migration. Fitness effects are the same as in Figure 3 (sf=0.05 throughout, sm=−0.4, 0, 1 in plots A–C). A deme number of n=400 was assumed. Each datapoint is based on
with sex-specific migration. Fitness effects are the same as in Figure 3 (sf=0.05 throughout, sm=−0.4, 0, 1 in plots A–C). A deme number of n=400 was assumed. Each datapoint is based on 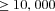 replicate simulations.
replicate simulations.
Overall, our results show that, in contrast to standard mitochondrial population genetics, the male migration rate can have an influence on the fixation probability of mitochondrial variants (although with the parameter combinations screened here this effect is only seen for very high rates of male migration). This effect arises because both male and female migration rates influence the inbreeding rate within demes and thus the degree to which male fertility influences effective selection on the mitochondrial mutation. In the extreme case where mm=1, all male offspring will leave their natal deme so that no inbreeding occurs within demes.
SIMULATION RESULTS: MIGRATION MODELS OTHER THAN WRIGHT’ ISLAND MODEL
So far, we have assumed Wright’s island model of migration only, in which migration is equally likely between all demes. To ascertain the robustness of our results with respect to this assumption, we obtained simulation results for two other, isolation-by-distance migration models: the stepping stone model (a one-dimensional string of demes connected by migration between adjacent demes) and the lattice model (two-dimensional version of the stepping stone model). In both cases, we assume that there are no border demes, that is, a ring or torus structure, respectively.
Our simulations indicate that the migration model has only weak effects on the fixation probability of mitochondrial mutations. For mutations that are neutral in females, the fixation probabilities of deleterious and neutral mutations in males do not depend on the migration model, as expected. For male beneficial mutations, increased population structure in the models of isolation by distance favors the spread of the mutation compared to the island model (Fig. 5) when the migration rate is high. This is expected from equation 1 because 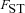 is higher in the isolation-by-distance models than in the island model. For low migration rates (
is higher in the isolation-by-distance models than in the island model. For low migration rates (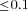 ), the fixation probabilities converge in the three models, as expected from the fact that
), the fixation probabilities converge in the three models, as expected from the fact that 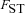 in models of isolation by distance converges to that of the island model (Rousset 2004). A slightly lower fixation probability can still be observed in the stepping-stone model at low migration rates and with very small demes (Fig. 5). Under such circumstances, stochastic deme extinctions occur throughout the population, which impedes the spread of the mitochondrial mutation more in the isolation-by-distance models than in the island model. This effect, however, is seen only for a restricted set of parameter values and for cases of extreme population structure with a large number of very small demes and limited female fecundity (e.g., 2N=10, n=1200, f=6, in Fig. 5). Fixation probabilities for the lattice model are always intermediate between those of the stepping-stone and island models.
in models of isolation by distance converges to that of the island model (Rousset 2004). A slightly lower fixation probability can still be observed in the stepping-stone model at low migration rates and with very small demes (Fig. 5). Under such circumstances, stochastic deme extinctions occur throughout the population, which impedes the spread of the mitochondrial mutation more in the isolation-by-distance models than in the island model. This effect, however, is seen only for a restricted set of parameter values and for cases of extreme population structure with a large number of very small demes and limited female fecundity (e.g., 2N=10, n=1200, f=6, in Fig. 5). Fixation probabilities for the lattice model are always intermediate between those of the stepping-stone and island models.

Estimates of the normalized fixation probability 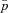 for a female neutral mutation (sf=0) with three different types of population structure: island model, stepping-stone model, and lattice model. Other parameters take the values m=0.1, n=1200, and N=5. Each datapoint is based on
for a female neutral mutation (sf=0) with three different types of population structure: island model, stepping-stone model, and lattice model. Other parameters take the values m=0.1, n=1200, and N=5. Each datapoint is based on 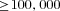 replicate simulations.
replicate simulations.
Discussion
Mutations in mtDNA that affect male fitness only are generally thought to be selectively neutral due to the maternal inheritance of mitochondria (Frank and Hurst 1996). Using both analytical methods and simulations, we have demonstrated here that mitochondrial mutations that reduce the fertility of males can be selected against when (1) the reduction in male fertility translates into a reduction in offspring number of the females that the male mates with, and (2) the population is subdivided into demes of small or moderate size. We will discuss both of these conditions in turn.
We have assumed that male fertility directly impacts offspring production: a female mating with a mutant male has (1+sm) times as many offspring as the same female mating with a wild-type male. Whether this is a realistic assumption will depend on the mating system as well as other factors. When females have only a single mating partner during their lifetime as assumed in our model, male fertility translates directly into female offspring production. Thus, this is the mating system where we can expect the strongest effects with respect to selection on mitochondria through their male fitness component. With sequential monogamy, male infertility may also directly impair offspring production. However, in this as in other systems where females mate with several males, females may also compensate to some extent for the low fertility of one of their mating partners. For example, females may save resources from producing fewer offspring in one breeding season, and then produce more offspring in the next breeding season when mating with a fully fertile male. Similarly, where females mate with multiple males in the same breeding season, they may compensate low sperm number or mobility from one male with normal sperm from another male. In the extreme case, a female that mates with both a mutant and a wild-type male may not suffer any reduction in her offspring production through infertility of the mutant male. In line with this reasoning, remating by a female has been shown to lead to increased offspring production in several species of Drosophila, which can be interpreted as an insurance against male sterility and subfertility (Singh et al. 2002). In spite of these and related effects, we expect the results of our model to be robust with respect to the details of the mating system as long as the term (1+sm) is interpreted not as the fertility of a male per se, but as the average change in offspring number of his female mating partners.
The second condition for selection on male-infertility mitochondrial mutations is that males carrying the mutation mate nonrandomly with females carrying the same mutation. The most straightforward scenario where this will happen is when females have a mating preference for related (inbreeding) or unrelated males (outbreeding or inbreeding avoidance). Inbreeding, modeled as nonrandom mating within a single population, has been shown to produce selection against male infertility mitochondrial mutations (Unckless and Herren 2009; Wade and Brandvain 2009), whereas outbreeding produces weak positive selection for such mutations (Engelstädter and Charlat 2006). In our model, inbreeding is a natural consequence of geographical population structure: with small demes and low migration rates, females are more likely to mate with relatives carrying the same mitotype than in a large panmictic population. As our results show, this can produce strong selection against male-infertility mutations even though mating is completely random within demes.
In contrast to male infertility mitochondrial mutations, population structure reduces the strength of selection on mutations that affect female fitness only. This is in accord with existing theory of nuclear genes (e.g., Maruyama 1970; Roze and Rousset 2003; Whitlock 2003). Putting the two components of selection through female and male effects together, it can be seen that population structure has a twofold effect on the fate of sexually antagonistic mutations: it reduces the efficacy of the female component but increases the efficacy of the male component of selection (see eq. 1). As a result, when both deme size and migration rate are sufficiently small, a large negative fitness effect on males can outweigh a small positive effect in females. Specifically, a female-beneficial but male-deleterious mutation will be selected against when the ratio of selection coefficients −sm/sf is larger than the number of females per deme (see eq. 3).
Mitochondrial mutations that impair male fertility have been reported in many animals as well as in humans (Gemmell et al. 2004; St. John et al. 2005; O’Flynn O’Brien et al. 2010). Because there is no direct selection on mitochondria through male fitness effects, this observation raises the question of what evolutionary forces keep these mutations in check. One obvious explanation is that these mutations are also deleterious in females. If these fitness effects in females are weak, these mutations may be maintained at a low frequency in the population through mutation-selection balance. Our model shows that in this case, population structure either weakens or strengthens selection against such mutations, depending on the migration rate (see Fig. 1C). A second explanation that applies for male infertility mutations that are neutral in females is that there will be strong selection at nuclear loci for compensatory mutations that ameliorate the male deleterious effect of the mitochondrial mutations. This notion is supported by reports of mitonuclear epistasis in animals (Rand et al. 2004; Dowling et al. 2008), and in particular the recent finding that mtDNA variation strongly impacts nuclear gene expression in male Drosophila, especially in the testes and accessory glands (Innocenti et al. 2011). Finally, our model offers the alternative explanation that population structure, acting in concert with deleterious effects of male infertility on female fecundity, can produce immediate selection against male infertility mitochondrial mutations that are neutral in females. Given that pronounced population structure as well as limited numbers of mates are common features of many species, we speculate that this effect could also contribute substantially to preventing mtDNA mutations from spreading. For humans, where ancient societies lived in small groups, this explanation might be especially attractive.
Associate Editor: N. Perrin
ACKNOWLEDGMENTS
We are grateful for L. Lehmann’s valuable suggestions for the problem of finite deme number populations. We would also like to thank D. Roze and an anonymous reviewer for helpful comments that substantially improved our manuscript. This work was supported by a scholarship from the China Scholarship Council to HZ and grants from the Swiss National Science Foundation to FG and JE (grant numbers PZ00P3_121697 and PZ00P3_132934).
Appendix
Here, we derive analytical expressions describing the spread of mitochondrial mutations in a limiting case of our model where there are many demes of finite size connected by migration according to Wright’s island model. The analysis below follows closely the derivations by Reuter et al. (2008) for microbes inducing cytoplasmic incompatibility, which in turn build upon previous work by Rousset (2004).
Let us denote by pij the frequency of the mutation in a particular female i in deme number j (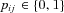 ). Furthermore, we define
). Furthermore, we define 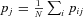 as the average frequency of the mutation among females in deme j, and
as the average frequency of the mutation among females in deme j, and 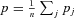 as the overall average frequency of the mutation among all females in the population. Likewise, we define qij, qj, and q as the frequency of the mutation in a particular male, among all males in a deme and among all males in the population. Because the mutation is maternally inherited and does not influence the mortality of males and females, we have p = q. We will, however, continue to use both terms wherever appropriate to distinguish between female and male effects in our notation.
as the overall average frequency of the mutation among all females in the population. Likewise, we define qij, qj, and q as the frequency of the mutation in a particular male, among all males in a deme and among all males in the population. Because the mutation is maternally inherited and does not influence the mortality of males and females, we have p = q. We will, however, continue to use both terms wherever appropriate to distinguish between female and male effects in our notation.
 ((A1))
((A1))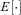 denotes the average over all demes in the population and all females within a deme.
denotes the average over all demes in the population and all females within a deme. ) and neglect terms
) and neglect terms 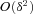 that are of order
that are of order 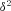 in the following analysis. For the fecundity of an individual female, we have
in the following analysis. For the fecundity of an individual female, we have
 ((A2))
((A2)) ((A3))
((A3)) ((A4))
((A4)) ((A5))
((A5))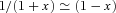 , equation (A5) becomes
, equation (A5) becomes
 ((A6))
((A6)) ((A7))
((A7))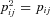 and p=q, and ignoring all terms of order
and p=q, and ignoring all terms of order 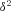 , we obtain after simplification
, we obtain after simplification
 ((A8))
((A8)) ((A9))
((A9)) ((A10))
((A10))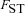 is the probability that the most recent common ancestor of two distinct individuals sampled from one deme also lived within that deme, and
is the probability that the most recent common ancestor of two distinct individuals sampled from one deme also lived within that deme, and 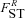 is the probability that two individuals sampled from a deme with replacement have their most recent common ancestor within that deme.
is the probability that two individuals sampled from a deme with replacement have their most recent common ancestor within that deme.


