

Up-regulation of the type 3 ryanodine receptor is neuroprotective in the TgCRND8 mouse model of Alzheimer’s disease
Abstract
The cellular pathology of Alzheimer’s disease is progressive and protracted leading eventually to considerable neuronal death. The underlying mechanisms of the pathology are complex but changes in the control of intracellular Ca2+ are believed to contribute to the demise of neurons. In this study, we investigated the functional consequences of an increase in the expression of the type 3 isoform of the ryanodine receptor (RyR3). We found that although cortical neurons from TgCRND8 mice secreted significantly more amyloid beta protein and showed significantly increased RyR3 expression, they were no more sensitive to cell stress than non-transgenic neurons. Furthermore, despite increased intracellular Ca2+ release in response to ryanodine, we found that basal Ca2+, K+-evoked Ca2+ responses, and capacitative Ca2+ entry were no different in TgCRND8 neurons compared with non-transgenic neurons. Therefore, as RyR3 up-regulation did not affect neuronal health or global Ca2+ homeostasis, we investigated the effect of reducing RyR3 expression using small interfering RNA. Surprisingly, a reduction of RyR3 expression in TgCRND8, but not in non-transgenic, neurons increased neuronal death. These data reveal a new role for RyR3 and indicate a novel potential therapeutic target to delay or prevent the progression of Alzheimer’s disease.
Abbreviations used:
-
- AD
-
- Alzheimer’s disease
-
- APP
-
- amyloid precursor protein
-
- Aβ42
-
- amyloid peptide
-
- [Ca2+]i
-
- intracellular calcium concentration
-
- calcein-AM
-
- calcein acetoxymethyl ester
-
- CICR
-
- Ca2+-induced Ca2+ release
-
- DIV
-
- days in vitro
-
- eGFP
-
- enhanced green fluorescent protein
-
- ER
-
- endoplasmic reticulum
-
- FAD
-
- familial AD
-
- MTT
-
- 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide
-
- PBS
-
- phosphate buffered saline
-
- PBST
-
- PBS containing 0.1% Tween20
-
- PS
-
- presenilin protein
-
- RyR
-
- ryanodine receptor
-
- RyR3
-
- type 3 isoform of the ryanodine receptor
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that is characterized at early stages by impairment of higher cognitive function and altered behavior (Jalbert et al. 2008). As the disease progresses, there is deposition of extracellular amyloid plaques and intracellular neurofibrillary tangles together with shrinkage of the neocortex because of extensive neuronal loss (Hyman and Trojanowski 1997). A highly penetrant occurrence of AD, termed familial AD (FAD) or early-onset AD, is a direct consequence of mutations in the genes that encode amyloid precursor protein (APP) and the presenilin proteins (PS1 and PS2). Since the pathogenesis of FAD and the more common sporadic AD are indistinguishable, mutant forms of APP and PS have been employed extensively to study the disease. There is now considerable evidence that APP and PS mutations increase the formation of the amyloid peptide, Aβ42, which compromises neuronal function and leads to neuronal death – the so-called ‘amyloid cascade hypothesis’ (Walsh and Selkoe 2007).
However, APP and PS mutations also affect the control of intracellular Ca2+ concentration ([Ca2+]i) (LaFerla 2002). Neuronal [Ca2+]i is precisely regulated by many different types of Ca2+ channels, Ca2+ pumps, and Ca2+ binding proteins, and a compromise in the function and/or expression of any of these components can disturb Ca2+ regulation and induce neuronal death (Berridge 1998). The hypothesis that Ca2+ dysregulation causes neuronal death in AD was postulated over a decade ago (Khachaturian 1994), but the precise involvement of Ca2+ dysregulation in AD pathogenesis is subtle, prolonged, and complicated (Green et al. 2007; Stutzmann 2007). For example, changes in the control of [Ca2+]i have been observed in various cell types, such as fibroblasts and platelets, even before cognitive decline and neuronal death occur in patients who develop AD (Etcheberrigaray et al. 1998). Furthermore, in several mice models that have mutations in APP and/or PS, Ca2+ release from the endoplasmic reticulum (ER) is potentiated but neuronal death is limited or absent (Stutzmann et al. 2006; Supnet et al. 2006). Clearly, the control of [Ca2+]i can change at a very early stage of AD, can occur in various cell types, remains modified for the duration of the disease, and yet does not necessarily result in overt cell death. Rather, Ca2+ dysregulation appears to increase the susceptibility of neurons to die as a result of other stressors such as glutamate and Aβ42 (Guo et al. 1999; Mattson et al. 2000).
It is interesting to note that, until recently, the presumed cause of Ca2+ dysregulation, and the associated increase in susceptibility of neurons to die, in FAD mutant transgenic mice was the result of increased Aβ42. This mechanistic interpretation arose because mutations in APP and PS increase the formation of Aβ42 and because various aggregated forms of Aβ42 applied directly to cultured neurons caused Ca2+ dysregulation and enhanced cell death (Mattson et al. 1992; Demuro et al. 2005; Supnet et al. 2006). However, many of these studies used transgenic mice expressing mutant versions of PS and recent data indicate that mutations in PS impact Ca2+ regulation in various ways independent of increased formation of Aβ42. For example, recent studies have revealed that PS can modulate capacitative Ca2+ entry (Leissring et al. 2000), may form ER Ca2+ leak channels (Tu et al. 2006), can modulate the gating of inositol trisphosphate receptors (Cheung et al. 2008), and can also modulate SERCA pump activity (Green et al. 2008).
We previously investigated Ca2+ dysregulation in cortical neurons from mice that have human APP with two FAD mutations (TgCRND8 mice, APP695 : KM670/671NL+ V717F; Supnet et al. 2006). These studies allowed an investigation of changes in the control of [Ca2+]i that result from misprocessing of APP but without the additional effects produced by mutations of PS. We found that cortical neurons from TgCRND8 mice exhibit enhanced Ca2+ release from the ER. Ca2+ is released from the ER through two types of Ca2+ channel – inositol trisphosphate receptors and ryanodine receptors (RyRs), both of which have three different isoforms encoded by different genes. We found that the increase in ER Ca2+ release in TgCRND8 neurons was a result of an increase in the expression of the type 3 isoform of the RyR (RyR3). Furthermore, we also demonstrated that an increase in expression of RyR3 in TgCRND8 neurons was a direct consequence of increased formation of Aβ42.
In this study, we investigated the effect of RyR3 up-regulation, and the consequent increase in ER Ca2+ release, on neuronal viability. Despite significant RyR3 up-regulation in cultured cortical neurons of TgCRND8 mice, there was no difference in susceptibility to cell death compared with non-transgenic neurons. There was also no significant difference in global Ca2+ signaling between TgCRND8 and non-transgenic neurons. However, and most strikingly, when we reduced the expression of RyR3 in TgCRND8 neurons using small interfering RNA (siRNA)-mediated knockdown, we observed an enhanced neuronal death. One possible mechanism by which RyR3 up-regulation may prevent neuronal death is by suppression of Aβ42 synthesis. However, we found no difference in concentration of Aβ42 and Aβ40 in the media of TgCRND8 neurons in which RyR3 was knocked down compared with untreated TgCRND8 neurons. These data reveal that up-regulation of RyR3 protects against, rather than contributes to, neuronal death in TgCRND8 cortical neurons. This is a novel role for RyRs and one that could potentially be exploited therapeutically to delay or prevent the progression of AD.
Materials and methods
Primary cortical cultures
Using methods described previously (Chan et al. 2000), cortical neurons were isolated from transgenic mice (TgCRND8) that express human APP with a double mutation: APP695 (KM670/671NL + V717F) (Chishti et al. 2001). Animal use was in accordance with the Canadian Council on Animal Care guidelines and approved by the Animal Care Committees of University of Prince Edward Island and National Research Council of Canada. Briefly, individual E16 fetuses were genotyped for the APP695 transgene by PCR and then all cortices of the same genotype were pooled, incubated with 0.25% trypsin in HEPES-buffered saline solution (Hyclone Logan, UT, USA) for 15 min and were dissociated using a large bore 1000 μL pipette tip. Cells were seeded onto poly-d-lysine-coated tissue culture plates or glass coverslips. Cell densities were 2 × 106 cells per well in six-well plates on 25 mm coverslips for long-term culture, 0.8 × 106 cells per well in 24-well plates on 13 mm glass coverslips for immunocytochemistry and Ca2+ imaging and 0.1 × 105 per well in 96-well plates for viability assays. Cultures were incubated at 37°C/5%CO2 in Neurobasal medium supplemented with B27 (Invitrogen, Carlsbad, CA, USA) and 10% fetal bovine serum (Hyclone) for 24 h, then cultured in serum-free Neurobasal medium supplemented with B27 thereafter. Half the conditioned media was replaced with fresh media every 3 days.
Viability assays
Calcein-AM assay
The viability of cortical cultures was quantified by calcein acetoxymethyl ester (calcein-AM; Invitrogen) uptake, the fluorescence of which is proportional to the number of living cells. Briefly, media were replaced with 200 μL/well phosphate buffered saline (PBS) containing calcein-AM (2 μg/mL). Plates were incubated for 45 min at 22°C in the dark and the fluorescence of each well was determined using a plate reader (SpectraMax M2; Molecular Devices, Sunnyvale, CA, USA). Percent cell viability for each condition was determined by averaging triplicate values and calculating the percentage change relative to the untreated condition.
MTT viability assay
The metabolic activity of cortical cultures was assessed using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT; Sigma-Aldrich, St. Louis, MO, USA) and this was used as an index of the number of living cells (Fig. S1a). For each well, half the media were replaced with PBS containing MTT (0.5 mg/mL) and the plates were incubated for 3 h at 37°C/5%CO2. After incubation, the culture media-MTT solution was removed and formazan crystals were dissolved in dimethylsulfoxide and the solution was transferred to a 96-well plate and read with a plate reader at 570 nm (SpectraMax M2). Percent cell viability for each condition was determined as for the calcein-AM assay.
NeuN viability assay
NeuN expression in cortical cultures was used as an indicator of neuronal viability where loss of NeuN staining indicated loss of neurons as a result of cell death. We confirmed that loss of NeuN immunofluorescence reports neuronal loss from culture by measuring the NeuN signal together with enhanced green fluorescent protein (eGFP) fluorescence from transfected neurons (tracking eGFP-positive neurons has previously been used to measure neuronal survival in vitro (Arrasate et al. 2004). We found a good correlation between loss of NeuN signal and loss of eGFP-positive neurons confirming that NeuN can be used to monitor neuronal death (Fig. S1b). Cells were fixed with PBS containing 4% paraformaldehyde for 15 min at 37°C/5%CO2, washed thrice with PBS and permeabilized with HEPES-Triton buffer for 10 min at 22°C. Cells were blocked with PBS containing 5% goat serum for 1 h at 22°C and incubated with NeuN primary antibody (1.25 μg/mL, Clone:A60; Chemicon, Temecula, CA, USa) in PBS containing 0.1% Tween20 (PBST) overnight at 4°C. Cells were returned to 22°C with three washes of PBS and then incubated in PBST containing AlexaFluor 488® (2 μg/mL; Invitrogen) for 1 h and then in PBST containing TO-PRO®-3 (1 μM; Invitrogen) for 15 min in the dark. Coverslips were mounted on slides using FluorSaveTM Reagent (Calbiochem, San Diego, CA, USA) and imaged by a researcher blind to the genotype and treatment condition. Approximately 10 images were acquired for each coverslip using an epifluorescence microscope (Axioskop 2 plus; Zeiss Canada, Toronto, ON) equipped with a digital camera (Axiocam MRm; Zeiss). All 12-bit images were acquired using Axiovision software (Zeiss) under identical conditions (2 s exposure, 10× objective and FITC filter set, Chroma, Rockingham, VA, USA; see Fig. S1c for representative images). NeuN signal in each image was quantified by image analysis (ImageJ, NIH). Using an image from the untreated condition (which contained the maximum number of living neurons), a threshold value was determined empirically. This was achieved by manually varying the threshold until only NeuN-positive nuclei were thresholded; the value of this threshold was then applied to all images in the experiment. The total pixel area captured by the threshold represented all NeuN-positive cells in the image and the total pixel area per image was used to represent the number of neurons alive at the time of fixation.
Single cell calcium imaging
[Ca2+]i in cortical neurons was measured as detailed previously (Bradley et al. 2006). Briefly, cells were loaded for 45 min at 37°C in HEPES-buffered saline (in mM: pH 7.4, 119 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 25 HEPES, and 30 glucose) containing fura-2AM (4 μM) and bovine serum albumin (0.1% w/v). Cells were washed with HEPES-buffered saline and incubated for a further 15 min. Individual coverslips were placed in a laminar flow chamber (FCS2; Bioptechs, Butler, PA, USA) and mounted on an inverted epifluorescence microscope (Axiovert 200; Zeiss) equipped with a CCD digital camera (Axiocam MRm; Zeiss). Neurons were perfused by gravity flow (2 mL/min), and precise switching between solutions was achieved with solenoid valves (Valvelink 8 controller; Automate Scientific, Berkeley, CA, USA). Slidebook software (Intelligent Imaging Systems, Denver, CO, USA) controlled excitation light from a Lambda DG-4 unit (Sutter Instrument, Novato, CA, USA) equipped to provide alternating excitation at 340 and 380 nm. Emitted light was collected with a fura-2 dichroic block (Chroma, Rockingham, VT, USA) at a rate of one image pair/2 sec. Image sequences were exported offline to ImageJ (NIH). Ca2+ responses were quantified by placing regions of interest on the soma of individual neurons and ratios of average intensities were calculated from pairs of 340/380 nm images at each time point. For the calculation of integrated Ca2+ responses for each neuron, a least-squares fit was applied to 10 points of the baseline and used to extrapolate a ‘predicted’ baseline for the next 20 points. The Ca2+ response was calculated as the sum of trapezoids at each time interval between the predicted baseline and the ratio values (Microsoft Excel).
siRNA knockdown of RyR3
siRNAs were generated using the Dicer siRNA Generation Kit (Genelantis, San Diego, CA, USA) as described previously (Supnet et al. 2006). Our previous protocol was modified to use LipofectAMINE since this transfection reagent produced no toxicity. Cortical cultures were transfected on day 1 as follows: culture medium was removed and the cells were washed twice with OptiMEM. A solution of LipofectAMINE in OptiMEM (0.1% v/v) containing either no siRNA, 200 ng siRNA to RyR3 or 200 ng control siRNA to GFP was added to the cells which were then incubated for 2 h at 37°C/5%CO2. Following incubation, the cells were returned to serum-free Neurobasal medium containing B27. Gene silencing was confirmed at 72 h and 14 days post-transfection.
RT-PCR detection and PCR quantification of RyR transcripts
Total RNA was extracted from 2 × 106 cortical cells using GenElute Kit (Sigma-Aldrich) and treated with DNase I. One μg total RNA was reverse-transcribed using the First Strand cDNA Synthesis Kit (Fermentas, Burlington, ON, Canada). Primers for RyR3 detection were specific to the region that was targeted by siRyR3 and yielded a PCR product of 613 base pairs; the primers were: 5′-TCATCTCTCGATATCGAATGG-3′ and 5′-ATGGAGTATGACCTTTC-3′. To quantify RyR3 mRNA expression after siRNA treatment, amplification of cDNA was conducted using an iCycler iQ Real-time PCR Detection System (BioRad Laboratories, Hercules, CA, USA). Primers were: actin, 5′-GGCTGTATTCCCCTCCATCG-3′ and 5′-TGTACCGTAACAATGGTTGACC-3′ and RyR3, 5′-ATCGCTGAACTCCTGGGTTTG-3′ and 5′-CCGATTCAAGGTAGCTGTACTT-3′. Relative gene expression across treatment conditions was calculated using the CT method (ΔΔCT), where the non-transgenic culture treated with siRNA to GFP was considered the control condition and 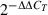 gave the relative expression of RyR3 mRNA.
gave the relative expression of RyR3 mRNA.
ELISA detection of Aβ42 and Aβ40
Media samples from cortical cultures were collected for the quantification of human Aβ using the hAmyloid β40/42 ELISA kit (Genetics Company, Schlieren, Switzerland) following the manufacturer’s instructions.
Statistical analysis
Data were analyzed using GraphPad Prism (version 3.02; Graphpad Software, San Diego, CA, USA). Two-way anova with Bonferroni post hoc test was used to determine the difference between genotypes and treatments and the possible interactions of each. Student’s t-test was applied when only two conditions were compared. To compare the distribution of [Ca2+] responses for populations of neurons cumulative frequency histograms were analyzed using the Kolmogorov–Smirnov test with a bin width of 2%. All differences were considered significant at p < 0.05.
Results
Cortical cultures from TgCRND8 mice are no more sensitive to cell death than those from non-transgenic mice
TgCRND8 cortical neurons show an up-regulation of RyR3 and an enhanced release of ER Ca2+ in response to glutamate and ryanodine (Supnet et al. 2006). Therefore, we considered the possibility that TgCRND8 neurons would be more sensitive to cell death or may die more pre-maturely as a result of dysregulated control of [Ca2+]i (LaFerla 2002). To test this hypothesis, both TgCRND8 and non-transgenic cortical cultures derived from the same cell preparation were treated with glutamate or H2O2. These cell stressors were added at 6 days in vitro (DIV) – a time in culture when there is a significant increase in the concentration of Aβ42 in the culture media and a significant increase in the expression of RyR3 in TgCRND8 cultures (Fig. S2a). As reported by others, we found that both glutamate and H2O2 induced a concentration-dependent loss of cells indicative of toxicity (Chan et al. 2000; Lovell et al. 2003) (Fig. 1a and b). However, TgCRND8 cultures did not show a significant difference in cell viability compared with non-transgenic cultures indicating no difference in the susceptibility to cell death. The percent survival following 24 h treatment with 100 μM glutamate was 52 ± 6.7% for non-transgenic cultures and 48 ± 4.2% for TgCRND8 cultures. The percent survival following 24 h treatment with 1 mM H2O2 was 23 ± 3.4% for non-transgenic cultures and 21.8 ± 2.3% for TgCRND8 cultures. Since apoptosis plays an important role in the pathophysiology of AD and since both RyRs and [Ca2+]i can trigger apoptotic signaling pathways (Lee et al. 2006), we determined if TgCRND8 neurons were more sensitive to cell death specifically by apoptosis. Using staurosporine, an inhibitor of protein kinase C, to induce apoptosis, we found that neuronal loss was not significantly different between TgCRND8 and non-transgenic cultures (Fig. 1c). The percent survival following 48 h treatment with 100 nM staurosporine was 57 ± 6.3% for non-transgenic cultures and 59 ± 6.8% for TgCRND8 cultures. Finally, a more subtle neuronal stress is aging and neurons in culture show a progressive decline in number as a result of cell death (Arrasate and Finkbeiner 2005). We therefore tested the effect of aging on the viability of TgCRND8 and non-transgenic cultures. Cortical cultures were grown for 18 DIV and cell viability was determined by MTT assay at the end of this culture period. Again, we found that TgCRND8 cultures were no more susceptible to cell death over time compared with non-transgenic cultures (Fig. 1d). Therefore, despite an up-regulation of RyR3 and an increase in the release of ER Ca2+ in TgCRND8 cultures, there does not appear to be an associated increase in the susceptibility for cells to die.

TgCRND8 cortical cultures are no more susceptible to cell death than non-transgenic cortical cultures. (a) Concentration-response of cortical cultures (6 DIV) to glutamate treatment (0–500 μM for 24 h). Cell viability was assessed with calcein-AM and fluorescence was read at 530 nm. (b) Concentration-response to H2O2 treatment (0–1000 μM for 24 h). Cell viability was assessed with the MTT assay and absorbance was read at 570 nm. (c) Concentration-response to staurosporine treatment (0–1000 nM for 48 h). Neuronal viability was determined by quantification of the NeuN intensity over an empirically determined threshold following immunocytochemistry. For each assay in a–c, cell viability is expressed as a percentage relative to the untreated condition. (d) MTT absorbance of cortical cells grown to 18 DIV. For all graphs, values are the mean ± SD of at least three independent experiments and no statistically significant difference was found between genotypes at any drug concentration as determined by anova.
Global Ca2+ responses are not changed in TgCRND8 neurons
Since we were unable to detect an increase in the susceptibility to cell death in TgCRND8 cultures, we considered whether the control of global [Ca2+]i was in fact affected as a result of up-regulation of RyR3. If, in TgCRND8 neurons, an increase in expression of RyR3 produces more functional Ca2+ channels – as we believe it does because of an enhanced release of ER Ca2+ in response to ryanodine (Supnet et al. 2006) – then one may anticipate potentiated global Ca2+ responses because of Ca2+-induced Ca2+ release (CICR). However, we found no significant difference in both resting Ca2+ concentration and K+-evoked global Ca2+ responses between TgCRND8 and non-transgenic neurons (Fig. 2a and b). Under basal conditions, the mean ratio of fura2 fluorescence at 510 nm when excited at 340 nm and 380 nm was 0.58 ± 0.18 for non-transgenic neurons and 0.62 ± 0.21 for TgCRND8 neurons. When all quantified global Ca2+ responses to K+ stimulation for each non-transgenic or TgCRND8 experiment (six cell preparations) were analyzed as cumulative frequency histograms, we found no significant difference in the population responses (Kolmogorov–Smirnov test, p < 0.05). Since mutations in PS were recently demonstrated to affect the SERCA pump (Green et al. 2008), we examined the rate of extrusion of Ca2+ from the cytoplasm following a K+ challenge. First-order exponential curve fits revealed no difference in the rate of removal of cytoplasmic Ca2+ in TgCRND8 and non-transgenic neurons. No significant difference was found when the mean half life of the curve fit was compared for responses from TgCRND8 (8.4 ± 2.3 s) and non-transgenic neurons (7.9 ± 2.5 s; Student’s t-test). Previous studies have also implicated mutations in PS in the control of capacitative Ca2+ entry. We therefore examined the Ca2+ response induced upon addition of extracellular Ca2+ to neurons previously treated with thapsigargin (to deplete ER Ca2+ stores and activate capacitative Ca2+ entry). Again, we found no significant difference between TgCRND8 (3.1 ± 1.18) and non-transgenic neurons (3.4 ± 2.3; Fig. 2d). Overall, these measurements revealed that although up-regulation of RyR3 potentiates release of Ca2+ from the ER in response to ryanodine and glutamate, other components of the Ca2+ signaling pathway were unaffected.

Global Ca2+ signaling is similar in neurons from TgCRND8 mice and those from non-transgenic mice. (a) Basal [Ca2+]i is not significantly different in TgCRND8 and non-transgenic neurons. Average 340/380 nm ratios were determined from a baseline of Ca2+ measurements of 10–15 frames. The graph shows mean ± SD for at least 50 neurons from six independent experiments. (b) The global Ca2+ responses to neuronal depolarization induced by K+ (45 mM for 30 s) are no different in TgCRND8 and non-transgenic neurons. The normalized cumulative frequency histogram (Norm. Cum. Frequency) was produced from the pooled data of six independent experiments. (c) The rate of extrusion of elevated Ca2+ following a K+ stimulus was not significantly different in TgCRND8 and non-transgenic neurons. Top panel shows a representative experiment of Ca2+ responses normalized to the first point of the Ca2+ decay. The black line is the average of all responses. Bottom panel shows mean ± SD of the half life derived from a single exponential curve fit of the Ca2+ response. The half life for the exchange of solutions in the laminar flow chamber was 0.6 s. (d) The size of capacitative Ca2+ influx is no different in TgCRND8 and non-transgenic neurons. Top panel shows representative traces of Ca2+ responses induced by the re-introduction of extracellular Ca2+ (black bar) after neurons had been treated with thapsigargin (1 μM for 5 min in the absence of extracellular Ca2+). The black line is the average of all responses. Bottom panel shows the mean ± SD of the quantified capacitative response (four independent experiments).
Knockdown of RyR3 induces cell death of TgCRND8 but not non-transgenic neurons
Since we were unable to detect any effect of RyR3 up-regulation on cell viability or Ca2+ signaling in TgCRND8 neurons, we considered two other possible consequences of RyR3 up-regulation: RyR3 up-regulation may have no functional consequence in neurons, or, conversely, RyR3 up-regulation may be a beneficial or compensatory function and therefore protect neurons. To test these ideas, we experimentally reduced RyR3 expression in both TgCRND8 and non-transgenic neurons using siRNA-mediated knockdown. Both TgCRND8 and non-transgenic cultures were transfected with either siRNA directed at RyR3 or with control siRNA directed at eGFP very soon after cell isolation (1 DIV; RyR3 up-regulation occurs as early as 6 DIV; Fig S2a). Knockdown of RyR3 mRNA was confirmed 72 h after transfection by RT-PCR (Fig. S2c). No change in the expression of RyR3 mRNA was detected with the control siRNA (Fig. S2c) and the expression of mRNA of RyR1 and RyR2 was the same in all conditions (data not shown).
We then determined if RyR3 knockdown affected the susceptibility of TgCRND8 and non-transgenic neurons to die when exposed to exogenous stressors. We found no significant difference in the susceptibility of either TgCRND8 or non-transgenic neurons to die when treated with glutamate or staurosporine 72 h after transfection with either siRNA to RyR3 or siRNA to eGFP (data not shown). We next examined the effect of RyR3 knockdown on the long-term survival of TgCRND8 and non-transgenic neurons. Since the neurons were cultured over 2 weeks following siRNA transfection, we performed quantitative PCR to determine if RyR3 remained knocked down 13 days after transfection (Fig. 3a). After culture for this duration, TgCRND8 neurons continued to show significantly greater expression of RyR3 than non-transgenic neurons (approximately 1.6-fold). However, the expression of RyR3 in TgCRND8 neurons transfected with siRNA to RyR3 remained significantly reduced (approximately sixfold). Interestingly, non-transgenic neurons treated with siRNA to RyR3 did not show a significant reduction in mRNA expression 2 weeks following siRNA transfection.

Knockdown of RyR3 induces cell death in TgCRND8 neurons but not in non-transgenic neurons. (a) Quantification of RyR3 mRNA expression in cortical cultures 13 days after siRNA transfection. mRNA expression was determined using quantitative RT-PCR and RyR3 expression was normalized to β-actin. All RyR3 mRNA expression was normalized to the non-transgenic culture treated with siRNA to GFP. Means ± SD are shown for six independent experiments. *p < 0.05, **p < 0.01; anova with post hoc Bonferroni test. (b) Quantification of neuronal number at different times (10, 14, and 18 days) in culture. TgCRND8 neurons treated with siRNA to RyR3 show a significant decrease in neuronal number at 18 DIV. Means ± SD are shown for data obtained from four independent experiments.
Having demonstrated prolonged knockdown of RyR3 mRNA in TgCRND8 neurons, we investigated cell viability. To specifically examine the effect of RyR3 knockdown on neuronal survival and to evaluate the time course over which cell death occurred, we performed immunocytochemistry using the neuron specific marker, NeuN, at 10, 14 and 18 DIV (Fig. 3b). As anticipated, there was a gradual, time-dependent loss of neurons in all treatment conditions. However, there appeared to be a greater decrease in the number of neurons in TgCRND8 cultures transfected with siRNA to RyR3 compared with control siRNA at 14 DIV and this difference was significant at 18 DIV. At 18 DIV, the average number of NeuN pixels per image for TgCRND8 neurons with the control siRNA was 25320 ± 1306 but this was significantly reduced to 11160 ± 1833 for TgCRND8 neurons with RyR3 knocked down. While neuronal death decreased with time in non-transgenic neurons, we found no significant effect of knocking down RyR3. Therefore, these data reveal that selective knockdown of RyR3 induces an accelerated rate of neuronal death only in neurons derived from TgCRND8 mice and not in non-transgenic neurons.
Knockdown of RyR3 does not affect the secretion of Aβ42 in TgCRND8 neurons
One possible mechanism by which a reduction of RyR3 expression may increase age-dependent neuronal death in TgCRND8 neurons is by increasing the secretion of Aβ42. If this was the case, then the up-regulation of RyR3 in TgCRND8 neurons may serve to suppress Aβ42 formation. Indeed, increases in cytosolic Ca2+ have been demonstrated to reduce the expression of Aβ42 and to increase the expression of soluble amyloid precursor protein-α (Buxbaum et al. 1994; Petryniak et al. 1996; Dreses-Werringloer et al. 2008). To test this hypothesis, we quantified the amount of Aβ42 secreted into the culture media by TgCRND8 neurons in which RyR3 had been knocked down and compared this with TgCRND8 neurons treated with control siRNA. We found no significant difference in the concentration of either Aβ42 or Aβ40 in TgCRND8 neurons with or without RyR3 knocked down (Fig. 4). After 72 h of siRNA treatment, the concentration of Aβ42 in the media of TgCRND8 neurons treated with siRNA to RyR3 was 13.4 ± 3.3 ng/mL and the concentration of Aβ42 in the media of TgCRND8 neurons treated with control siRNA was 15.0 ± 0.3 ng/mL. We also found no significant difference in the concentration of Aβ40 which was 17.8 ± 4.9 ng/mL and 14.9 ± 0.9 ng/mL in TgCRND8 neurons treated with siRNA to RyR3 and control siRNA, respectively.

Knockdown of RyR3 in TgCRND8 neurons does not affect the synthesis of Aβ42. Cortical neurons from TgCRND8 and non-transgenic embryos were cultured for 5 days and then transfected with either control siRNA or siRNA for RyR3. After 72 h, media were removed and concentration of both Aβ42 and Aβ40 was determined by ELISA. Knockdown of RyR3 in TgCRND8 neurons treated with siRNA RyR3 was confirmed by RT-PCR. Mean ± SD are shown and were obtained from four independent experiments.
Discussion
In this study, we analyzed cortical neurons from TgCRND8 embryos and found that, despite a significant increase in the expression of an ER Ca2+ channel – the type 3 ryanodine receptor (RyR3) – we did not observe an associated compromise in neuronal health. Furthermore, up-regulation of RyR3 did not affect basal [Ca2+]i, nor did it affect global Ca2+ responses to K+ depolarization. However, a reduction of RyR3 expression in TgCRND8 neurons by siRNA-mediated knockdown increased neuronal death. This increased neuronal death was not a result of increased formation of Aβ42. These data reveal that up-regulation of RyR3 may provide a neuroprotective role in cortical neurons from TgCRND8 mice. Although the mechanism by which this protective effect is afforded is not yet known, potentiation of RyR3 function appears to be a plausible therapeutic strategy for future investigation.
RyR3 up-regulation, Ca2+ dysregulation, and neurotoxicity
Dysregulation of Ca2+ homeostasis has, for a long time, been implicated in neuronal death in both AD and normal aging (Toescu and Verkhratsky 2007; Bezprozvanny and Mattson 2008). It is therefore interesting that we unable to demonstrate an increase in susceptibility to neuronal death in TgCRND8 neurons which have significantly increased expression of RyR3 and which release significantly more ER Ca2+ when treated with ryanodine or glutamate (Supnet et al. 2006). Most studies using FAD transgenes have analyzed neurons derived from mutant PS mice or mice that have multiple FAD transgenes including a mutant PS transgene. The presence of mutant PS is important for the interpretation of these experiments because mutations in PSs do not just affect the processing of APP to increase the ratio of Aβ42/Aβ40, but have additional adverse effects. For example, PS in the γ-secretase complex cleaves other substrates in addition to APP, such as Notch, and as such the misprocessing of multiple γ-secretase substrates may affect neuronal viability independent of Ca2+ signaling (Selkoe and Wolfe 2007). PSs have also been reported to form Ca2+ leak channels in the ER membrane and mutations may affect the steady state ER Ca2+ concentration (Tu et al. 2006; Nelson et al. 2007). Therefore, an increased susceptibility to neuronal death induced by PS mutations may result from the modification of multiple cellular processes in addition to the proteolytic cleavage of APP. Furthermore, mouse models that have only mutations in APP and not in PS do not exhibit extensive neuronal death (Chishti et al. 2001; Hsia et al. 1999) Since we used neurons from TgCRND8 mice, our experiments were performed in the context of wild-type PSs and therefore were unaffected by additional adverse changes that occur with PS mutations. Using TgCRND8 neurons, we previously demonstrated that enhanced formation of Aβ42 increases RyR3 expression and enhanced ER Ca2+ release. However, we now report that these changes are insufficient to increase the susceptibility of these neurons to die.
Impact of RyR3 up-regulation on Ca2+ signaling
As a result of increased RyR3 expression, and associated increase in ER Ca2+ release, one may anticipate greater than normal Ca2+ signals because of increased CICR. However, when we tested this hypothesis, we found no difference in global Ca2+ signals when neurons were depolarized with a submaximal concentration of K+. These findings are similar to those of Stutzmann et al. (2006) who noted that despite enhanced Ca2+ responses to uncaged InsP3 in neurons from a triple transgenic mouse, they found no difference in the global Ca2+ responses induced by action potential generation. A probable explanation for these findings is that the potentiation of CICR by RyR up-regulation is too small to change global Ca2+ responses in neurons. It is also important to note that in non-transgenic cortical neurons, RyR3 expression is much lower than that of RyR1 and RyR2. As such, the increase in RyR3 expression in TgCRND8 cortical neurons contributes only a small percentage of the total RyR expression in these neurons. Our inability to detect global Ca2+ responses in TgCRND8 neurons is probably the result of this relatively small increase in total RyR expression. However, spatially restricted changes in the expression of RyR3 may potentiate subcellular CICR and enhance Ca2+ responses in a spatially and temporally defined manner. Indeed, Stutzmann et al. recently demonstrated that Ca2+ responses to ryanodine are more greatly potentiated at the Schaffer collateral-CA1 synapse than those in the soma of CA1 neurons of the triple transgenic mice (Chakroborty et al. 2009). Interestingly, the work of Chakroborty et al. (2009) reported an increase in expression of the type 2 RyR, not the type 3 RyR as we report. Although we do not have a clear explanation for this difference, it is important to note that we measured RyR mRNA from cultured cortical neurons whereas Chakroborty et al. measured RyR mRNA from hippocampal samples.
A number of studies have revealed that PS mutations affect capacitative Ca2+ entry, a mechanism essential for the refilling of intracellular Ca2+ stores. We found no difference between TgCRND8 and non-transgenic neurons when capacitative Ca2+ entry was evoked. Further, we found no difference in the rate of extrusion of intracellular Ca2+ following a large global Ca2+ response (induced by K+ depolarization). Therefore, despite an increased potential to release ER Ca2+ in TgCRND8 neurons, other properties of Ca2+ handling were unaffected.
RyR3 up-regulation and neuroprotection
Having established that RyR3 up-regulation in TgCRND8 neurons does not affect neuronal viability, we considered the effect of reducing the expression of up-regulated RyR3 in TgCRND8 neurons. Surprisingly, early and sustained knockdown of RyR3 in TgCRND8 neurons lead to increased neuronal death. However, this enhanced neuronal death was only detectable when a subtle stress, that of long-term in vitro culture, was applied to the neurons, and not when an acute stress of exogenous stressor (glutamate and staurosporine) was applied. Furthermore, the enhanced neuronal death with long-term culture was not immediate. In fact, significantly greater neuronal death was only observed 14 days after knockdown of RyR3 (assuming knockdown of RyR3 protein 3 days after transfection with siRNA). We believe that the absence of an increase in sensitivity to exogenous stressors upon knockdown of RyR3 in TgCRND8 neurons reveals something of the nature of the effect of RyR3 expression changes in TgCRND8 neurons. We believe the reduction in RyR3 expression subtly compromises cell viability that only becomes evident over an extended time in culture. Acute stress conditions to the neurons appear to be too severe to reveal the subtle deleterious effect of reduced RyR3 in TgCRND8 neurons. In addition, acute changes in [Ca2+]i in TgCRND8 neurons in which RyR3 was knocked down were not observed (basal Ca2+ and K+-evoked responses; data not shown). Therefore, we do not believe that gross changes in Ca2+ handling are the cause of death in TgCRND8 neurons when RyR3 is knocked down.
It is interesting to note that RyR3 mRNA is induced in TgCRND8 neurons within 4 days of culture and yet neuronal death resulting from the reduction of RyR3 expression was not significantly different from the control siRNA until 2 weeks later. A number of scenarios could explain the delay in neuronal death in TgCRND8 neurons following RyR3 knockdown. First, RyR3 up-regulation may only be one of a number of cellular responses that act to preserve neuronal viability in neurons expressing a mutant human APP transgene. As such, the compromised total cellular compensatory response produced by RyR3 knockdown may only fail, and so lead to cell death, when the toxic insult becomes more severe as may occur with increased time in culture. Second, although RyR3 expression is increased very soon after exposure of neurons to Aβ42 (24 h; Supnet et al. 2006), an increase in RyR3 expression may only be protective for neurons at advanced age in culture, possibly only when they are fully differentiated. Finally, it is also important to note that knockdown of RyR3 did not compromise neuronal viability in non-transgenic neurons. We believe that this indicates that RyR3 does not, under normal physiological conditions, subserve a protective role. As such RyR3 appears to be recruited as a compensatory response as a result of expression of the human APP transgene in TgCRND8 neurons. Although we do not yet know the mechanism by which RyR3 up-regulation contributes to a neuroprotective compensatory response, we anticipate from the data of this study that facilitation of RyR3 function may delay the adverse effects of Aβ42.
Acknowledgements
We are very grateful for the gift of the TgCRND8 mice from Dr David Westaway (University of Alberta, Alberta, Canada). This research was supported by an industry award from the Alzheimer’s Society of Canada, the Canadian Institutes for Health Research (CIHR), AstraZeneca and the National Research Council of Canada. CS was supported by a Doctoral Research Award from CIHR and a Graduate Student Award from the Scottish Rite Charitable Foundation of Canada.

