

An aging-related signature predicts favorable outcome and immunogenicity in lung adenocarcinoma
Wenjing Zhang and Yuting Li contributed equally to this work.
Funding information
This study was supported by the Shandong Provincial Youth Innovation Team Development Plan of Colleges and Universities (No. 2019-6-156, Lu-Jiao), National Natural Science Foundation of China (Nos. 81872719 and 81803337), Provincial Natural Science Foundation of Shandong Province (No. ZR201807090257), and National Bureau of Statistics Foundation Project (No. 2018LY79)
Abstract
Aging has been demonstrated to play vital roles in the prognosis and treatment efficacy of cancers, including lung adenocarcinoma (LUAD). This novel study aimed to construct an aging-related risk signature to evaluate the prognosis and immunogenicity of LUAD. Transcriptomic profiles and clinical information were collected from a total of 2518 LUAD patients from 12 independent cohorts. The risk signature was developed by combining specific gene expression with the corresponding regression coefficients. One cohort treated with the immune checkpoint inhibitor (ICI) was also used. Subsequently, a risk signature was developed based on 21 aging-related genes. LUAD patients with low-risk scores exhibited improved survival outcomes in both the discovery and validation cohorts. Further immunology analysis revealed elevated lymphocyte infiltration, decreased infiltration of immune-suppressive cells, immune response-related pathways, and favorable ICI predictor enrichment in the low-risk subgroup. Genomic mutation exploration indicated the enhanced mutation burden and higher mutation rates in significantly driver genes of TP53, KEAP1, SMARCA4, and RBM10 were enriched in patients with a low-risk signature. In the immunotherapeutic cohort, it was observed that low-risk aging scores were markedly associated with prolonged ICI prognosis. Overall, the estimated aging signature proved capable of evaluating the prognosis, tumor microenvironment, and immunogenicity, which further provided clues for tailoring prognosis prediction and immunotherapy strategies, apart from promoting individualized treatment plans for LUAD patients.
Abbreviations
-
- COSMIC
-
- Catalogue of Somatic Mutations in Cancer
-
- GEO
-
- Gene Expression Omnibus
-
- GSEA
-
- gene set enrichment analysis
-
- ICI
-
- immune checkpoint inhibitor
-
- LUAD
-
- lung adenocarcinoma
-
- LUSC
-
- lung squamous cell carcinoma
-
- NB
-
- neoantigen burden
-
- NSCLC
-
- non–small-cell lung cancer
-
- SMGs
-
- significantly mutated genes
-
- ssGSEA
-
- single sample GSEA
-
- TCGA
-
- The Cancer Genome Atlas
-
- TCIA
-
- The Cancer Immunome Atlas
-
- TMB
-
- Tumor mutation burden
1 INTRODUCTION
Based on data obtained from the latest cancer statistics report, NSCLC is the major cause of cancer-related morbidity and mortality.1 It primarily comprises 2 histology subtypes, namely, LUAD and LUSC.2 Amongst these, LUAD accounts for approximately 55% of all NSCLC patients.3 Although improved clinical treatment strategies, such as combined treatment and targeted therapy (eg, tyrosine kinase inhibitors), have been developed for LUAD, the 5-y survival rate remains quite poor.4, 5 Therefore, more effective and robust indicators with respect to prognosis evaluation are urgently needed to identify patients who are suited to distinct medication programs.
The emergence of ICI therapy has dramatically prolonged the survival outcomes of several advanced cancers, including melanoma,6 renal cancer,7 and NSCLC.8 Moreover, ICI treatment has become a pillar of LUAD clinical practice, alongside conventional surgery, chemotherapy, and targeted treatment. At present, blockading the immune checkpoints of programmed cell death protein 1 (PD-1) or its ligand PD-L1 is the best described immunotherapy method9 and is fast becoming the routine first-line treatment strategy for NSCLC patients.10 Despite the marked therapeutic advantage of ICI agents observed in both clinical trials and real-world populations, the biggest limitation of this treatment modality is that only a subset of patients could obtain clinical benefits from such immune treatment.11 Therefore, novel immunotherapeutic efficacy determinants that could be used for selecting patients who are suitable for ICI treatment need to be identified.
Age is a well known risk biomarker for most diseases, including human cancers. In addition to age itself, its related genomic molecular features have also been revealed to be associated with disease progression and survival. Several recent studies have reported that specific genes (eg, APOE12 and FOXO312, 13), genomic regions (eg, 5q33.314), and multiple single-nucleotide polymorphisms15, 16 are linked to longevity. It is difficult to dissect and investigate aging because of its complex interactions with numerous factors, including genome, environment, and age-relevant diseases.17 To comprehensively depict the transcriptomic landscape of aging, Peters et al17 conducted a population-based large-scale transcriptomic analysis and determined aging-related genes.
In this study, in total, 2518 LUAD cases from 12 independent cohorts were curated from publicly available data sources to construct and validate an aging-related risk signature. To investigate the potential mechanisms underlying the identified risk signature, multi-dimensional immunology analyses were conducted, and the results demonstrated the significant capabilities of this signature in evaluating the tumor microenvironment. Findings gleaned from our work may provide helpful clues for prognosis assessment and immunotherapy prediction in LUAD patients.
2 MATERIALS AND METHODS
2.1 Publicly available LUAD cohorts, immunotherapy cohort, and aging-relevant genes
The GEO and TCGA databases were searched to obtain eligible LUAD samples with both gene expression profiles and clinical characteristics. Samples without transcriptome data/follow-up information or those diagnosed as other lung cancer types were excluded from the candidate list. Finally, in total, 2518 samples were acquired for subsequent analysis, including TCGA (N = 499), GSE72094 (N = 398), GSE68465 (N = 442), GSE50081 (N = 127), GSE42127 (N = 132), GSE41271 (N = 181), GSE31210 (N = 226), GSE30219 (N = 85), GSE13213 (N = 117), GSE26939 (N = 115), GSE11969 (N = 90), and GSE81089 (N = 106). GSE50081, GSE31210, and GSE30219 were integrated as the GEO-meta cohort 1 due to the fact that they shared the same microarray platform (ie, Affymetrix Human Genome U133 Plus 2.0 Array). Similarly, GSE42127 and GSE41271, which were both detected using the Illumina HumanWG-6 v3.0 expression BeadChip, were combined into the GEO-meta cohort 2. The ComBat function embedded in the R sva package18 was applied to eliminate batch effects from distinct cohorts. Among the included LUAD datasets, as TCGA had the largest sample size with 499 patients, it was regarded as the discovery cohort and used for establishing the risk signature. The detailed dataset information and microarray platforms of all the LUAD samples are shown in Table S1. In total, 348 urothelial cancer samples treated with the anti-PD-L1 agent (ie, atezolizumab) in the IMvigor210 cohort19 were curated to further investigate the relationship between the identified risk signature and immunotherapy prognosis. The complete clinical features, survival information, and gene expression profiles of the immunotherapy cohort were acquired from http://research-pub.gene.com/IMvigor210CoreBiologies. A comprehensive list of 1433 aging-related genes was obtained from a recently published study,17 that provided a transcriptional landscape of age.
2.2 Construction of the aging-related risk signature
All 1433 aging-related genes were subjected to univariate Cox regression analysis based on the gene expression data of the LUAD discovery cohort to identify the genes involved in prognosis risk. Thereafter, prognosis-related aging genes were subjected to the Lasso-Cox regression model (implemented by R glmnet package20) to determine the genes that contributed the most to survival. According to the Lasso coefficient profile, the optimal gene combination can be identified to construct the risk signature. By combining specific gene expression levels with their corresponding regression coefficients weighted by the Lasso analysis, risk cores for each LUAD patient could be calculated. The detailed formula was as follows: risk score = 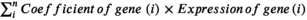 . All patients were stratified into low-risk and high-risk subgroups with the median risk score as the cutoff value. Based on this risk stratification, the association of the determined aging risk signature with LUAD survival outcomes was evaluated in both the discovery and validation cohorts.
. All patients were stratified into low-risk and high-risk subgroups with the median risk score as the cutoff value. Based on this risk stratification, the association of the determined aging risk signature with LUAD survival outcomes was evaluated in both the discovery and validation cohorts.
2.3 Estimation of tumor-infiltrating immune cells and immune checkpoints
To elucidate the distinct immune infiltration levels in low-risk vs high-risk subgroups, evaluation of the abundance of 28 tumor-infiltrating immune cell phenotypes, which were recently reported by Charoentong et al21 study was performed. The 28 immunocytes were divided into the following 3 categories to analyze their distinct immune functions: anti-tumor, pro-tumor, and intermediate tumor immunocytes. The feature genes for each immune cell subtype are listed in Table S2. Moreover, the CIBERSORT method is capable of calculating the abundance of 22 human lymphocyte subtypes with 547 feature genes from the leukocyte gene signature matrix, named LM22.22 The present study used both approaches to obtain mutual-validation results.
In addition, a complete list of immune checkpoint genes was collected from a previously published immunogenomic study.23 In the discovery LUAD cohort, the checkpoint gene VISTA was not observed due to the diverse sequencing platform. Therefore, we compared the distinct expression of 33 genes in the low-risk and high-risk subgroups.
2.4 Quantification of the enrichment of immunotherapy and immunogenicity predictors
Several recently reported immune-related signatures have been demonstrated to be associated with immunogenicity and immunotherapy responses. Therefore, this study curated 6 representative signatures as follows: (a) T cell-inflammation signature, which is composed of 18 genes involved in a T cell-activated tumor microenvironment and linked with efficacy to pembrolizumab24; (b) cytolytic activity25; (c) immune cell signature, which indicates the overall immunocyte infiltration level in the microenvironment26; (d) cytokines and chemokines27; (e) immune signaling molecules27; and (f) immune cell subsets, which reflect the infiltrating abundance of T cells, B cells and natural killer (NK) cells.27 The gene panels used for evaluating the enrichment scores of each immune signature are presented in Table S3.
2.5 GSEA and ssGSEA
GSEA was applied to infer the dysregulated pathways of distinct risk subgroups. The t values extracted from the differential analysis results of genome-wide expression (performed by the limma package28) were regarded as the input variable for the ‘fgsea’ function implemented by the R fgsea package (https://github.com/ctlab/fgsea). Subsequently, signaling pathways derived from the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) databases were used as annotation circuits. The enriched pathway plot was obtained using the clusterProfiler package.29 To evaluate the enrichment of specific gene panels involved in distinct immune cell subtypes and signatures, the single sample GSEA (ssGSEA) method within the GSVA package30 was used to calculate the enrichment scores of each signature for each LUAD sample.
2.6 Extraction of the mutational signatures operative in the genome
The SignatureAnalyzer software31 was used for the detection of mutational signatures present in the genome, on the basis of mutation profiles from TCGA LUAD cohort. Bayesian non-negative matrix factorization is the core algorithm of SignatureAnalyzer and was used to optimally determine the number of mutational signatures. Specifically, mutation portrait matrix A was decomposed into 2 non-negative matrices W and H (ie, A ≈ W × H), in which W reflects the identified mutational signatures and H indicates the corresponding mutational activities. The determined mutational signatures were subsequently compared with the 30 well annotated signatures derived from the COSMIC database,32 according to the coefficient of cosine similarity.
2.7 Identification of the SMGs
The MutSigCV algorithm33 was used to identify SMGs in LUAD samples. One SMG could be included in the final list if it met the following 3 criteria: statistically significant, expressed in TCGA LUAD, and an encyclopedia of cell lines.34 According to the principle proposed by the Kandoth et al35 study, a gene was considered to be expressed if it had 3 or more reads in 75% or more of the samples. Mutational patterns of the identified SMGs in distinct subgroups were illustrated using the maftools package.36
2.8 Acquisition and definition of the mutational burden
TMB was regarded as the total nonsynonymous mutation count per megabase based on somatic mutation data obtained from TCGA cohort. In addition, the NB was defined as the total neoantigen count of each sample. The neoantigen data of 486 LUAD samples were acquired from TCIA project (https://www.tcia.at/patients). The detailed neoantigen calculation is shown in Supplementary Methods. Taking into account the crucial roles of DNA maintenance genes (eg, TP53, POLE, and BRCA1/2) in genomic stability, the mutations of the above genes were incorporated into the multivariate logistic regression model to adjust the confounding factors and obtain a real connection of the constructed aging signature with the mutational burden.
2.9 Statistical analysis
R software (version 4.0.2) was used to perform the related analyses. Heatmap exhibition of determined aging genes in distinct risk subpopulations was completed using the pheatmap package. Moreover, the Kaplan-Meier approach was applied to draw survival curves, and the significant difference was assessed via the log-rank test. Multivariate regression models within the forestmodel package were used to rule out biased factors and to acquire a controlled link. The relationship of continuous and categorical variables with 2 risk subgroups was evaluated using the Wilcoxon rank-sum test and Fisher exact test, separately.
3 RESULTS
3.1 Identification of the aging risk signature
As mentioned above, in total, 2518 LUAD patients were acquired after excluding samples without the requisite transcriptome expression data or prognosis information. As the LUAD cohort from TCGA harbors the largest sample size (N = 499) and detailed clinicopathologic characteristics, it was regarded as the discovery cohort to determine the aging risk signature that is linked with the survival outcome and immune infiltration of LUAD patients. Univariate Cox regression analysis of the curated 1433 aging-related genes was performed using gene expression profiles from TCGA cohort. The results suggested that 256 genes were prognostic (all P < .05; Table S4). Subsequently, the Lasso-Cox regression model was used with 10-fold cross-validation to determine the aging genes with the greatest contribution to LUAD prognosis. The Lasso coefficient profile between the log (λ) and the gene combination number is illustrated in Figure 1A. It was observed that the minimized partial likelihood deviance could be obtained when the combination number was 21 (Figure 1B). Therefore, we selected 21 aging-related genes to construct a risk signature for LUAD prognosis evaluation.

The aforementioned 21 genes comprised ZNF101, PLEKHB1, P2RX1, EIF2AK3, LPAR6, ATF7IP2, MS4A1, CCR2, ZNF10, AKTIP, GNG7, DAAM2, PTTG1, IL1R2, KYNU, TCN1, ITGA6, AHSA1, DSC1, LINGO2, and C1QTNF6. The survival contribution coefficients for LUAD patients in TCGA cohort are depicted in Table S5. The present study established a risk signature to evaluate the risk score for each LUAD patient (Figure 1C) based on the linear integration between the identified 21 gene expression values and their corresponding regression coefficients obtained via the Lasso-Cox model. The risk association plot of the evaluated risk scores, along with survival times and outcomes, is shown in Figure 1C. The expression levels of the identified 21 genes in distinct risk subgroups have also been illustrated with a heatmap (Figure 1C).
To explore the prognostic ability of the constructed risk signature, LUAD patients from the discovery cohort were classified into low-risk (N = 249) and high-risk (N = 250) subgroups with the median risk value as the cutoff value. It was observed that low-risk patients harbored a significantly improved survival outcome compared with the high-risk patients (log-rank test P < .001; Figure 1D). This association remained significant even after adjusting for age, sex, stage, and smoking status in the multivariate Cox regression model (HR: 0.32, 95% CI: 0.23-0.46, P < .001; Figure 1E).
3.2 Validation of the aging risk signature
To corroborate the prognostic roles of the determined aging signature, this study used 8 other LUAD cohorts. In 4 GEO cohorts of GSE72094, GSE68465, GEO-meta cohort 1, and GEO-meta cohort 2, it was observed that patients with low-risk signature scores had significantly prolonged survival outcomes compared with high-risk patients (log-rank test P < .001 for GSE72094, P = .008 for GSE68465, and P < .001 for GEO-meta cohorts 1 and 2; Figure 2A,C,E,G). Multivariate Cox regression models with clinical confounders (eg, age, sex, stage, grade, and smoking status) taken into consideration further confirmed the independent prognostic efficacy of the aging risk signature (GSE72094, HR, 0.42, 95% CI, 0.28-0.63, P < .001; GSE68465, HR, 0.71, 95% CI, 0.54-0.93, P = .012; GEO-meta cohort 1, HR, 0.54, 95% CI, 0.37-0.79, P = .002; GEO-meta cohort 2, HR, 0.49, 95% CI, 0.33-0.73, P < .001; Figure 2B,D,F,H). Moreover, clinical correlation analysis revealed that the elevated signature risk scores were significantly enriched in the LUAD patients with advanced clinical stage (TCGA, GSE72094, GEO-meta cohorts 1 and 2, all P < .05; Figure S1A–D) or worse histological grade (GSE68465 cohort, Kruskal-Wallis H test, P < .001; Figure S1E).

In the remaining GEO LUAD cohorts of GSE13213 (N = 117), GSE26939 (N = 115), GSE11969 (N = 90), and GSE81089 (N = 106), the results demonstrated that the trends of preferable survival outcomes were observed in patients with lower risk scores, although they did not attain statistical significance (log-rank test, all P > .05; Figure S2). This may be attributed to the relatively small sample size.
3.3 Correlation of the identified aging signature with favorable immune infiltration and tumor immunogenicity
Recently, several studies have revealed that aging and its related molecular features are associated with cancer immune response and regulation.37, 38 Therefore, it was hypothesized that the identified risk signature may modulate immunocyte infiltration and signaling pathways related to immunogenicity. A heatmap based on the ssGSEA method was constructed to visualize the distinct infiltrating abundance of 28 lymphocyte subtypes between the low-risk and high-risk subgroups in the discovery cohort (Figure 3A). The results indicated that anti-tumor immunocyte subtypes, such as activated CD8 T cells, effector memory CD8 T cells, NK cells, and type 1/17 T helper cells were markedly infiltrated in the low-risk signature LUAD patients (all P < .05). Consistently, decreased infiltration of immune-suppressive lymphocytes, such as CD56 dim NK cells and type 2 T helper cells was observed in the low-risk subgroup (both P < .05). In addition, the infiltration of activated/immature B cells, eosinophils, mast cells, monocytes, and T follicular helper cells, which belong to the intermediate immunocyte group, was also significantly enriched in the low-risk patients. Moreover, an immune infiltration analysis was also performed using the CIBERSORT algorithm, and similarly, the preferable lymphocyte infiltration represented by CD8 T cells was noticed in the low-risk group (Figure S3).

GSEA against KEGG/GO databases and LUAD transcriptomic expression profiles was applied to explore the signaling circuits associated with the aging risk signature. Consequently, it was observed that the immune response-related pathways in the KEGG database (eg, T cell receptor signaling pathway, cytokine receptor interaction, and chemokine signaling pathway) and GO database (eg, activation of immune response, adaptive immune response, antigen receptor mediated signaling pathway, and B cell receptor signaling pathway) were significantly present in LUAD patients with low-risk scores (Figure 3B,C).
Several molecular signatures concerning immunotherapy efficacy, immunogenicity, as well as immune cell status, were curated and their distinct enrichment was evaluated according to risk subgroups. Patients with low-risk signature scores harbored significantly elevated enrichment of the T cell-inflammation signature, cytolytic activity, and immune cell signature compared to high-risk patients (Wilcoxon rank-sum test, all P < .01; Figure 3D–F). In addition, a markedly higher enrichment of cytokine/chemokine signature, immune signaling molecules, and immune cell subset signature was observed in this low-risk subgroup (Wilcoxon rank-sum test, all P < .01; Figure S4).
The distinct expression of comprehensive immune checkpoint genes in the low-risk and high-risk subgroups was assessed. The results demonstrated that most of the genes (eg, PD-L1, PD-1, and CTLA-4) exhibited enhanced expression in patients with the low-risk aging signature (all P < .05; Figure S5).
3.4 Genomic features associated with the identified aging signature
The genomic mutational burden has been demonstrated to play a vital role in cancer progression and clinical immunotherapy response.39, 40 Therefore, the present study evaluated the association of the aging risk signature with TMB and NB. Based on the genomic mutation data obtained from TCGA cohort, we calculated the TMB for each LUAD sample and observed that patients with a low-risk signature had a significantly elevated TMB compared with the high-risk group (Wilcoxon rank-sum test, P = .008; Figure 4A). Subsequently, using the neoantigen data from the TCIA project, a consistent result between the low-risk signature and higher NB was observed (Wilcoxon rank-sum test, P = .021; Figure 4B).

Mutational signatures, which are characterized by distinct combinatorial patterns of nucleotide substitutions, have been shown to contribute to mutation burden and immune treatment outcomes. Therefore, we extracted potential mutational signatures present in LUAD samples by decomposing the nucleotide substitution matrix using the non-negative matrix factorization (NMF) method. According to the cophenetic metric profile, 3 mutational signatures were found to potentially exist (Figure 4C). After comparing them with the well validated signatures in the COSMIC database and using the cosine similarity (Figure 4D), 3 mutational signatures were finally determined as signature 1 (initiated by spontaneous deamination of 5-methylcytosine), signature 2 (attributed to the activity of the AID/APOBEC family), and signature 4 (associated with smoking). Their distinct mutation patterns are depicted in Figure 4E. Three mutational signature activities across all LUAD patients were evaluated and are presented in Figure 4F and Table S6. Further analysis revealed that low-risk signature scores were markedly correlated with increased mutational activities of signature 1 (P = .029; Figure S6A) and decreased activities of signature 2 (P = .008; Figure S6B). No significant difference was observed between the 2 risk subgroups in relation to signature 4 (P = .412; Figure S6C).
To investigate whether the higher TMB of low-risk patients was influenced by other confounding factors, clinical characteristics (ie, age, sex, stage, and smoking status), extracted mutational signatures (ie, signatures 1, 2, and 4), and mutations in genome maintenance regulators (eg, POLE, BRCA1/2, and TP53) were incorporated into the multivariate logistic regression model. To this end, the association of the low-risk aging signature with the increased TMB remained significant (OR: 1.87, 95% CI: 1.08-3.28, P = .028; Figure 4G).
In total, 23 SMGs were identified based on the genomic mutational profile of the discovery cohort. The SMG waterfall plot of low-risk vs high-risk subgroups (Figure 5) revealed a significantly distinct mutation rate in TP53 (104 of 247 [42.1%] vs 131 of 248 [52.8%]; P = .019), KEAP1 (31 of 247 [12.6%] vs 58 of 248 [23.4%]; P = .002), SMARCA4 (13 of 247 [5.2%] vs 33 of 248 [13.3%]; P = .003), and RBM10 (12 of 247 [4.9%] vs 24 of 248 [9.7%]; P = .048). The association between low-risk patients and lower TP53 mutation frequency was also observed in 2 validation cohorts (GSE72094: 17.3% vs 30.9%, P = .002; GSE13213: 20.7% vs 44.8%, P = .009; Figure S7A,B). Nevertheless, in the GSE11969 cohort with the smallest sample size (N = 90), no significant difference in TP53 mutation rate was observed between the 2 risk subpopulations (26.7% vs 37.8%, P = .367; Figure S7C). Other SMG mutation data were unavailable for the above 3 LUAD validation datasets.

3.5 The identified aging risk signature for evaluating immunotherapy prognosis
Considering that the constructed aging signature was demonstrated to be associated with tumor microenvironment and immunogenicity, it was hypothesized that this risk signature may contribute to the ICI prognosis. Therefore, an advanced urothelial cancer anti-PD-L1 immunotherapeutic cohort (Imvigor210) with transcriptome data and clinical information was used to explore the association of the identified risk signature with ICI response and outcome. The results suggested that patients with low-risk aging signature scores exhibited a significantly improved ICI survival outcome compared with patients with high-risk scores (log-rank test P = .017; Figure 6A). Moreover, this association remained significant after adjusting for sex, ECOG score, smoking history, and immune phenotype in the multivariate Cox regression model (HR: 0.75, 95% CI: 0.57-0.97, P = .028; Figure 6B). Further analysis demonstrated that more ICI responders were enriched in the low-risk subgroup (30.0% vs 16.5%, P = .006; Figure 6C). In addition, an elevated proportion of patients with disease control was also found in the low-risk group (52.1% vs 36.7%, P = .009; Figure 6D). Finally, an immunology analysis was performed with the gene expression profile of this ICI cohort. Consistent with the aforementioned results, a significantly increase in the enrichment of immune-related signatures (all P < .01; Figure 6E), immune response-relevant signaling pathways from KEGG and GO databases (Figure 6F,G), as well as favorable immune cell infiltration (Figure S8) were observed in patients with the low-risk aging signature.

4 DISCUSSION
By integrating transcriptomic data and clinical information from multiple independent LUAD cohorts, this study developed and verified an aging-related risk signature for survival prediction and immunogenicity evaluation. A forte of our study is that the risk signature was constructed based on a larger LUAD population and validated with distinct dimensions. Further prospective research is necessary, but the results from this work indicate the potential roles of the aging risk signature in LUAD survival outcome, immune microenvironment, and ICI treatment efficacy.
Mutational signatures are the fingerprints of endogenous and exogenous factors that represent distinct mutational patterns.41 The aging-related mutational signature 1 was recently reported to be associated with weaker immune infiltration and worse survival outcomes in triple-negative breast cancer and prostate cancer,42 suggesting its potential implication in immune treatment response. Furthermore, Chong et al43 leveraged genomic mutation data from melanoma and NSCLC patients treated with ICI agents and observed that patients with this aging mutational signature exhibited an inferior prognosis in both tumors. In the present study, distinct from mutation-specific indicators, an aging-related risk signature was developed based on the transcriptomic level, and it was elucidated that this signature was predictive of survival status and immune infiltration.
Higher TMB, NB, and immunogenicity were demonstrated to be associated with better survival outcomes in both early and late NSCLC, although the use of TMB for early NSCLC is considered controversial. To rule out the possibility that the improved LUAD outcome was induced by the above factors, multivariate Cox regression analyses were performed with these factors taken into account (Figure S9A), along with interaction analyses of the aging signature with TMB, NB, and 6 immune-related signatures using Cox regression (Figure S9B), as well as mutational burden stratification analyses (Figure S9C–F) using TCGA cohort. The associations between the aging signature and improved LUAD outcomes derived from the above analyses were consistent. These results suggested that the determined aging risk signature is an independent prognostic biomarker for LUAD patients.
Of the 21 identified aging-related genes, 4 (ie, P2RX1, CCR2, PTTG1, and MS4A1) were also found to be linked to cancer immune response or evasion. Wang et al44 discovered that a subset of P2RX1-deficient neutrophils was an immunosuppressive factor that induces the evasion of anti-tumor immunity in metastatic pancreatic tumors. The CCR2 deletion in cancer cells was established to associate with decreased tumor proliferation and improved survival in a mouse model, which was due to the enhanced infiltration of cytotoxic T cells and upregulation of MHC-I members.45 Furthermore, a recent study reported that PTTG1 knockdown could increase the ionizing radiation-triggered T cell immune response via the TGF-β1/SMAD3 circuit and further ameliorate the immunosuppressive tumor microenvironment.46 The elevated expression of MS4A1 was demonstrated to be associated with a favorable prognosis as well as efficacious anti-PD-1 treatment, due to its positive association with T cell immunity.47 These findings further support the predictive role of our constructed risk signature in immunocyte infiltration and ICI responses.
Based on recent findings, worse clinical outcomes were observed in lung cancer patients with mutations in TP53,48 KEAP1,49 SMARCA4,50 or RBM10.51 In this study, the low-risk LUAD patients harbored lower mutational frequencies of the above 4 genes, which was consistent with the association between low-risk scores and improved prognosis. Nonetheless, several studies have also reported the positive roles of TP53 and SMARCA4 mutations in evaluating favorable ICI treatment responses.50, 52, 53 High-risk patients with elevated mutation rates of TP53 and SMARCA4 nevertheless exhibited inferior ICI outcomes, indicating that more immunosuppressive factors (eg, CD56dim NK cells and type 2 T helper cells) were present in this subgroup, which inhibited the ICI promotion roles derived from the gene mutations. Therefore, further in-depth studies are required to achieve a conclusive understanding of the same.
To elucidate the clinical implications of identified aging signature in LUAD, the present study examined its association with well known prognostic or immunologic indicators. Taking into consideration the significant correlations between this signature and multiple indicators, it is proposed that the aging risk signature may be a potential biomarker for evaluating LUAD prognosis and immunogenicity. Nevertheless, there are certain disadvantages posed by previously reported indicators. For instance, the crucial roles of TMB have been demonstrated in cancer prognosis and immune response.54, 55 However, different sequencing platforms, lack of accurate threshold definition, and whole-exome sequencing costs hinder the broad utilization of TMB in clinical practice.56 For another biomarker, PD-L1, its high protein expression has been reported to be associated with a better immunotherapy response in NSCLC.57, 58 However, some patients with PD-L1-negative cancer also exhibited a favorable treatment response.59 These findings suggest that novel and robust biomarkers are necessary, and our established aging signature may be a potential candidate for LUAD patients.
There are several limitations to this study. First, the transcriptomic gene expression data of LUAD patients were obtained from publicly accessible cohorts, which may introduce biases in data analysis between distinct datasets. Second, the somatic mutational data were used only from TCGA cohort and lacked relevant mutation-level validation. Moreover, the potential mechanisms underlying the connections between the aging risk signature and multiple immunologic indicators remain unclear and require further experimental validation. Finally, the identified aging signature was associated with survival outcomes in both the non-ICI as well as the ICI cohorts. This suggests that this signature might solely be a prognostic biomarker, rather than a predictive biomarker. Therefore, LUAD cohorts with both transcriptomic data and immunotherapy information as well as clinical trials are necessary to further validate the ICI predictive roles of the aging signature.
In summary, using the aging-related transcriptomic profiles based on a larger LUAD population, the present study developed a risk signature to assess survival outcome, immunogenicity, and ICI treatment efficacy. The identified aging risk signature may be considered a potential biomarker for LUAD prognosis evaluation and immunotherapy response prediction.
ACKNOWLEDGMENTS
QHW thanks WJZ for her company over the past 9 y and for giving a cute baby.
DISCLOSURE
The authors have no conflict of interest.
Open Research
DATA AVAILABILITY STATEMENT
All data used in this study are acquired from publicly available cohorts.

