

Diversity and linkage disequilibrium in farmed Tasmanian Atlantic salmon
Summary
Farmed Atlantic salmon (Salmo salar) is a globally important production species, including in Australia where breeding and selection has been in progress since the 1960s. The recent development of SNP genotyping platforms means genome-wide association and genomic prediction can now be implemented to speed genetic gain. As a precursor, this study collected genotypes at 218 132 SNPs in 777 fish from a Tasmanian breeding population to assess levels of genetic diversity, the strength of linkage disequilibrium (LD) and imputation accuracy. Genetic diversity in Tasmanian Atlantic salmon was lower than observed within European populations when compared using four diversity metrics. The distribution of allele frequencies also showed a clear difference, with the Tasmanian animals carrying an excess of low minor allele frequency variants. The strength of observed LD was high at short distances (<25 kb) and remained above background for marker pairs separated by large chromosomal distances (hundreds of kb), in sharp contrast to the European Atlantic salmon tested. Genotypes were used to evaluate the accuracy of imputation from low density (0.5 to 5 K) up to increased density SNP sets (78 K). This revealed high imputation accuracies (0.89–0.97), suggesting that the use of low density SNP sets will be a successful approach for genomic prediction in this population. The long-range LD, comparatively low genetic diversity and high imputation accuracy in Tasmanian salmon is consistent with known aspects of their population history, which involved a small founding population and an absence of subsequent introgression. The findings of this study represent an important first step towards the design of methods to apply genomics in this economically important population.
Advancements in sequencing technologies have dramatically improved the availability and power of genomic tools for the study of important agriculture production species. Salmonid researchers now have access to a high quality reference genome assembly (Lien et al. 2016) and SNP arrays to collect genotypic data across populations (Kent et al. 2009; Gidskehaug et al. 2011; Barson et al. 2015; Yáñez et al. 2016). These tools have propelled two broad areas of research which will benefit the salmon farming industries. The first is identification of functional mutation(s) within genes that confer major effects on economic traits using genome-wide association studies (GWAS). A powerful recent example is the identification of VGLL3 and its effect on age at maturity (Ayllon et al. 2015; Barson et al. 2015). The second area concerns the implementation of genomic prediction, whereby genotypic data are used to estimate the genetic merit of breeding candidates. This has proven to be highly successful in dairy cattle (Hayes et al. 2013), and early studies in salmon are also positive (Odegård et al. 2014; Tsai et al. 2015). Both approaches are influenced by the strength of linkage disequilibrium (LD) that persists between markers within the population under investigation. Specifically, the extent of LD impacts the power to detect QTL associations in GWAS and the ability to successfully tag SNP effects and impute missing genotypes during the estimation of genomic breeding values (Kemper & Goddard 2012). As a precursor to both GWAS and genomic prediction within Atlantic salmon in Tasmania, this study sought to evaluate levels of genetic diversity and LD using a genome-wide collection of SNPs.
Tasmania, located off the southern part of mainland Australia, is not part of the natural range of Atlantic salmon. Efforts to introduce and acclimatise them to Australia commenced in the 1860s with sea voyages from England carrying ova collected from rivers in Scotland, England and Wales (Clements 1988). Repeated early efforts to establish a breeding population were unsuccessful, and today's Tasmanian farmed stocks originate from the River Philip in Nova Scotia, Canada. A total of 782 individuals were sampled from the selective breeding program managed by Salmon Enterprises of Tasmania, an Australian commercial hatchery and smolt producer. This population, abbreviated as ‘TAS’ throughout, contains pedigreed individuals from 11 year classes spanning 2001 to 2011. Samples were stored as fin clips under ethanol at ambient temperature until DNA extraction before genotyping was performed using a custom 220 000 SNP Affymetrix array developed by AquaGen (Norway) and the Centre of Integrative Genetics (CIGENE, Norway) and described elsewhere (Barson et al. 2015). Genotyping was performed via a collaboration with CIGENE, generating data from a total of 218 132 variants. Data sub-setting to isolate particular SNP collections or groups of individuals was performed using the suite of manipulation tools implemented within plink v1.9 (Purcell et al. 2007). Data filtering included the removal of five individuals with a SNP call rate < 99% and 656 SNPs with a call rate < 90%. The genotypic dataset is available from the Dryad repository with DOI 10.5061/dryad.pm354. Existing genotypes were obtained from two wild populations of European Atlantic salmon assayed using the same SNP array. The two populations, referred to throughout as FIN55 and FIN56, were collected from the Tana main stream in northern Finland and are described in detail elsewhere (Barson et al. 2015).
Levels of genetic diversity within the TAS, FIN55 and FIN56 populations were assessed using four diversity metrics. Genotypes from each population were used separately to estimate the proportion of polymorphic SNPs (PN), average gene diversity (HE) and average pairwise genetic distance separating individuals (DST) using plink v1.9. As shown in Table 1, the proportion of loci that displayed polymorphism was markedly different, with only around half of tested SNPs displaying both alleles within the TAS animals (PN = 0.537). Expected heterozygosity was much lower in the TAS animals compared to either European wild population when calculated using all SNPs (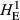 ) or only the subset of population-specific polymorphic loci (
) or only the subset of population-specific polymorphic loci (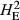 ). The final metric was DST, which reports the average genetic distance separating pairs of animals drawn from within each population. This revealed that the TAS individuals were more closely related to each other (DST = 0.2) than to animals within either of the wild populations (DST = 0.313 and 0.310 respectively; Table 1). To assess the allele frequency spectrum of 106 492 polymorphic SNPs, minor allele frequencies (MAFs) were estimated using all 777 fish. The distribution of MAFs is given in Fig. 1a and revealed a strong skew towards lowly polymorphic loci. The highest proportion of loci (23%) was found in the lowest MAF category (<0.05). The excess of SNPs with a low rate of polymorphism was extreme, with more than 55% of polymorphic loci having MAFs lower than 0.15. This distribution was compared with the allele frequencies observed within 137 wild European salmon using the same collection of SNPs (Fig. 1b). In these animals, only 14% of the loci displayed low MAF (<15%) and the majority of SNPs had MAFs between 20% and 35%. This distribution is similar to that observed using a QC filtered set of approximately 130 000 loci genotyped in farmed populations from Scotland and Norway (Houston et al. 2014). This suggests that the distribution seen here in wild fish is a product of their European origin rather than because of differences between wild and farmed populations.
). The final metric was DST, which reports the average genetic distance separating pairs of animals drawn from within each population. This revealed that the TAS individuals were more closely related to each other (DST = 0.2) than to animals within either of the wild populations (DST = 0.313 and 0.310 respectively; Table 1). To assess the allele frequency spectrum of 106 492 polymorphic SNPs, minor allele frequencies (MAFs) were estimated using all 777 fish. The distribution of MAFs is given in Fig. 1a and revealed a strong skew towards lowly polymorphic loci. The highest proportion of loci (23%) was found in the lowest MAF category (<0.05). The excess of SNPs with a low rate of polymorphism was extreme, with more than 55% of polymorphic loci having MAFs lower than 0.15. This distribution was compared with the allele frequencies observed within 137 wild European salmon using the same collection of SNPs (Fig. 1b). In these animals, only 14% of the loci displayed low MAF (<15%) and the majority of SNPs had MAFs between 20% and 35%. This distribution is similar to that observed using a QC filtered set of approximately 130 000 loci genotyped in farmed populations from Scotland and Norway (Houston et al. 2014). This suggests that the distribution seen here in wild fish is a product of their European origin rather than because of differences between wild and farmed populations.
| Population | SNPs | n | P N |
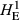
|
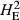
|
D ST |
|---|---|---|---|---|---|---|
| FIN55 | 208 704 | 137 | 0.999 | 0.381 | 0.381 | 0.313 |
| FIN56 | 208 704 | 326 | 0.999 | 0.380 | 0.380 | 0.310 |
| TAS | 218 132 | 782 | 0.537 | 0.119 | 0.222 | 0.200 |
-
The number of loci (SNPs) and number of individuals (n) is given for each population. Estimates of within-population diversity were measured as the observed proportion of polymorphic SNPs (PN), and expected heterozygosity measured using all available SNPs (
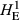 ) or the population specific subset of SNPs showing polymorphism (
) or the population specific subset of SNPs showing polymorphism (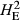 ). These were estimated using the Hardy-Weinberg equilibrium flag implemented in plink v1.9. The average pairwise genetic distance separating individuals is given as DST, where increasing values indicate an elevated genetic distance separating fish.
). These were estimated using the Hardy-Weinberg equilibrium flag implemented in plink v1.9. The average pairwise genetic distance separating individuals is given as DST, where increasing values indicate an elevated genetic distance separating fish.
- FIN55 and FIN56, two populations of Atlantic salmon collected from the Tana main stream in northern Finland; TAS, the population of 782 individuals sampled from the selective breeding program managed by Salmon Enterprises of Tasmania.

The main objective of the study was to evaluate the strength and persistence of LD within the TAS population. LD was estimated for SNP pairs using r2, which is less prone to overestimation compared with alternative metrics such as D′ (Ardlie et al. 2002). LD was calculated for loci pairs separated by up to 500 kb using --r2 --ld-window-kb 500 in plink v1.9. The base pair coordinate for each SNP was taken from salmon reference genome version ICSASG_v1, available from the European Nucleotide Archive as assembly GCA_000233375.3. Base pair positions allowed examination of the relationship between r2 and the physical distance separating SNP pairs. Loci separated by short distances (<50 kb) retained extensive LD in the TAS population with average r2 values ranging between 0.3 (at 50 kb) up to 0.67 for SNP pairs closer than 1 kb apart (Table 2). The average value of r2 in 1-kb intervals is plotted in Fig. 1c. As expected from other species, LD declined in a smooth curve with increasing physical distance. At longer physical distances (>150 kb), detectable and decreasing average r2 was observed out to the maximum interval evaluated (500 kb; Table 2). The values and decline of this decay curve were compared to wild European fish. Using the same set of 106 492 SNPs, LD at short distances was low (<0.05) and quickly dissipated to background levels. For example, the average LD in the FIN population for markers separated by 0–10 kb was only 0.037, which is an order of magnitude lower than for the farmed Tasmanian population (Table 2). It is not surprising to observe low LD in a wild and outbred population such as the FIN salmon; however, it is more surprising to observe such extensive LD as contained in the TAS salmon. The results are consistent with a pilot study of the TAS population that used many fewer loci (Dominik et al. 2010). Comparison with published estimates in other populations of Atlantic Salmon are difficult to directly compare, as they have been reported in units of recombination (Gutierrez et al. 2015) or sliding windows of 20 SNPs (Johnston et al. 2014). In both cases, the authors discuss the generally low levels of LD encountered, suggesting less extensive LD than encountered in the TAS population. Our observations concerning the lower polymorphism rate, skewed MAF and likely ascertainment bias prompted re-estimation of r2 using a subset of SNPs selected to contain higher MAFs. This sought to explore if the extensive LD observed was strongly influenced by using a biased excess of loci displaying low allele frequencies. A subset of 21 372 SNPs was defined that displayed high MAFs (>0.3) in the TAS population and had an average nearest neighbour distance of 200 kb (Table S1). Re-estimation of LD using this SNP set returned r2 values similar to the full set of polymorphic loci. The decay curve drops slightly faster at short LD, however, in general is not meaningfully different (Fig. 1c). This suggests that the largest factor contributing to the extensive LD observed is not the ascertainment of SNPs used to measure it. Finally, we examined the effect of year class stratification by estimating average r2 using animals drawn from the largest year class represented (2008, 123 animals). Average r2 was slightly higher (Table S2), reflecting elevated relatedness between animals within the same generation; however the increase was small.
| Marker distance (kb) | Tasmanian population | Finnish population | ||||
|---|---|---|---|---|---|---|
| All fish | Males | Females | All fish | |||
| 106K SNPs | 21K SNPs | 106K SNPs | 106K SNPs | 106K SNPs | 21K SNPs | |
| 0–10 | 0.540 | 0.441 | 0.541 | 0.541 | 0.037 | 0.032 |
| 10–20 | 0.412 | 0.361 | 0.414 | 0.414 | 0.027 | 0.025 |
| 20–30 | 0.363 | 0.327 | 0.366 | 0.366 | 0.024 | 0.022 |
| 30–40 | 0.334 | 0.303 | 0.335 | 0.335 | 0.022 | 0.021 |
| 40–50 | 0.312 | 0.285 | 0.314 | 0.314 | 0.021 | 0.020 |
| 50–100 | 0.270 | 0.248 | 0.272 | 0.272 | 0.019 | 0.017 |
| 100–200 | 0.211 | 0.215 | 0.212 | 0.213 | 0.017 | 0.016 |
| 200–300 | 0.171 | 0.177 | 0.173 | 0.173 | 0.016 | 0.014 |
| 300–500 | 0.131 | 0.153 | 0.133 | 0.133 | 0.015 | 0.014 |
- Average linkage disequilibrium values (r2) were calculated for marker pairs within the distance intervals given at the far left (in kb). Values were estimated using either 106 492 SNPs found to be segregating in the Tasmanian population (106K SNP) or a subset of 21 372 found to have high minor allele frequency (MAF <0.3) in the same population (21K SNP). Values were compared using all fish in the Tasmanian sample (n = 777), the males only (n = 277) and the females only (n = 254) or the FIN55 population, which was used to represent European Atlantic salmon (n = 137). Background LD was estimated by randomly sampling 1000 SNPs and averaging r2 among 476 603 non-syntenic marker pairs. The resulting value (r2 = 0.009) was below the average values obtained for markers separated by up to 500 kb, suggesting weak LD persists across even larger chromosomal distances.
To begin assessing the prospects for the performance of genomic prediction in the TAS population, we tested the accuracy with which genotypic data could be imputed. SNPs were first excluded that were either monomorphic or in complete LD (r2 > 0.99) with another locus on the same chromosome. This identified a set of 78 925 SNPs. The population of 777 animals was split into two panels: (i) a reference panel consisting of 574 fish from year classes 2001 to 2008, which was used to build a collection of observed haplotypes using all SNPs, and (ii) a test panel consisting of 203 fish from the 2009 to 2011 cohorts with simulated low density SNP data. To simulate four low-density SNP sets within the test panel, all genotypes were converted to missing except for 500 (0.5K set), 999 (1K set), 3036 (3K set) or 4933 loci (5K set). The SNPs with retained genotypes were the 158th (0.5K set), 79th (1K), 36th (3K) or 16th (5K) marker based on chromosomal position, creating sets of evenly distributed loci. A family-based method for imputation was applied, as this reflects the likely implementation within industry when pedigree data is available. Missing genotypes were imputed using fimpute 2.2 default parameters (Sargolzaei et al. 2014), which exploits family data and has been shown to produce accuracies equivalent to or higher than beagle in less time (Piccoli et al. 2014). The results showed that high levels of imputation accuracy were obtained. Imputation up to 78 925 loci using the 5K SNP set was achieved with 0.9667 accuracy. This means the proportion at which directly observed vs. imputed genotypes differed was around 3%. The observed accuracies decreased using lower density SNP sets as follows: 0.9588 accuracy using the 3K set, 0.9245 using the 1K set and 0.8910 using the 0.5K set. It is worthwhile noting that imputation using only 500 SNPs represents reliance on less than 1% of available loci.
The key findings in this study are the low level of genetic diversity and extensive LD that persists in farmed Tasmanian Atlantic salmon. The low diversity likely results in part from ascertainment bias in the SNP array used to measure it, as the 220K array is enriched for variants discovered using European strains (Barson et al. 2015). The observed long-range LD, however, is less likely to be strongly influenced by SNP ascertainment (see Fig. 1c). A much more plausible explanation can be found in the population history shaping Tasmanian Atlantic salmon, which is characterised by introductions from a single geographic source (one Canadian river) with no subsequent introgression from divergent strains since. The expected consequences are consistent with the reduced genetic diversity, elevated genetic similarity between individuals and long-range LD observed. This has implications for the application of GWAS and genomic prediction for industry. The high LD observed using only 20 000 loci means that genome scans using medium density platforms (tens of thousands of loci) are likely to have high power to detect gene effects of moderate to large size. Regarding implementation of genomic prediction, we found that imputation accuracies higher than 95% were achieved using as few as 3036 loci. This suggests that low-density panels will be sufficient to capture and tag the collection of population haplotypes, providing promise that progress can be realised for complex traits relevant to industry such as growth rate, product quality and disease resistance. In summary, documenting the decreased level of genetic diversity and behaviour of LD using over 100 000 polymorphic loci represents an important preliminary step towards speeding genetic gain in this important aquaculture species.
Acknowledgements
We thank the efforts of Thomas Moen (AquaGen), Sigbjorn Lien, Matthew Kent, Harald Grove (CIGENE) and colleagues for the developmental work resulting in construction and availability of the Affymetrix 220K array used in this work. The authors have no conflicts of interest.

