

Lp(a) and Risk of Recurrent Cardiac Events in Obese Postinfarction Patients
Abstract
Studies of recurrent coronary events in obese postinfarction patients show mixed results despite potential importance of obesity-related pathophysiologic processes and associated markers in establishing and predicting risk. The study aim was to determine specific markers of recurrent risk in obese postinfarction patients. Nondiabetic patients of the Thrombogenic Factors and Recurrent Coronary Events (THROMBO) postinfarction study were classified according to BMI as normal weight (<25 kg/m2), overweight (25.0–29.9 kg/m2), and obese (≥30 kg/m2). Cox multivariable regression with adjustment for significant clinical covariates was performed in each group monitoring outcome (cardiac death, myocardial infarction (MI), or unstable angina with 26 months follow-up) as a function of 17 thrombogenic, inflammatory, and metabolic blood markers and 17 cardiovascular disease-associated genetic polymorphisms. Results revealed no statistically significant genetic or blood marker variables in normal or overweight patients. For obese postinfarction patients, elevated lipoprotein(a) (Lp(a))was found to be a highly significant risk marker with hazard ratio and 95% confidence interval of 3.94 (2.11–7.35), P = 0.000017 (upper tertile vs. lower two tertiles). Additionally, elevated Lp(a) was found to interact with the −75G>A polymorphism of the apolipoprotein A-I gene and the −250G>A polymorphism of the hepatic lipase gene in establishing risk. We conclude that interactions of elevated Lp(a) with polymorphisms of the apolipoprotein A-I and hepatic lipase genes, primarily reflective of altered lipoprotein metabolism, play an important role in the establishment of recurrent coronary event risk in obese, nondiabetic postinfarction patients. These findings suggest close monitoring and consideration of weight reduction for obese postinfarction patients with elevated Lp(a) levels.
Introduction
Obesity is an independent risk factor for development of cardiovascular disease (CVD) (1); however, the role of obesity as a risk factor for recurrent coronary events in patients with established CVD is less clear (2,3,4,5,6,7,8,9,10). A number of studies in patients with previous myocardial infarction (MI) or other forms of stable CVD show independent association of increased BMI with various outcome measures including reinfarction, cardiac death, all-cause death, coronary revascularization, and stroke (2,3,4,5,6,7), while other studies show no association of BMI with cardiac death in postinfarction or poststroke patients (8), no association of BMI with reinfarction or cardiac death in postinfarction patients (9), and inverse association of BMI with all-cause death in postinfarction patients (9,10).
In addition to lack of definitive epidemiologic demonstration of obesity as a risk factor in patients with stable CVD, little is known regarding potential mediation of risk in such patients by specific obesity-related mechanisms, although both insulin resistance and low-grade inflammation (dyslipidemia, adipokine production, procoagulation, hypofibrinolysis, oxidative stress, endothelial dysfunction) have been thought to be important in this regard (11). Thus, it follows that obese patients with stable CVD might have different risk factors for recurrence than nonobese patients with stable CVD. To test this hypothesis, we performed Cox multivariable regression analysis of outcomes (re-infarction, cardiac death, or unstable angina—whichever occurred first) in nondiabetic patients of the Thrombogenic Factors and Recurrent Coronary Events postinfarction study (12) stratified by BMI and adjusting for significant clinical covariates as a function of 17 thrombogenic, inflammatory, and metabolic blood markers and 17 CVD-associated genetic polymorphisms.
Methods and Procedures
Study population
The study population comprised nondiabetic patients (N = 845) of the Thrombogenic Factors and Recurrent Coronary Events study and was carried out with approval of and according to guidelines of the Research Subjects Review Boards of participating centers. Enrollment eligibility included patients of either sex who were ≥21 years of age and admitted to 13 participating hospitals for MI. Exclusions included coronary bypass graft surgery during hospitalization; significant comorbidity such as malignancy or severe hepatic, renal, or cerebral disease; or heparin-type anticoagulant therapy. Overweight and obesity were defined as BMI of 25.0–29.9 kg/m2 and BMI ≥ 30 kg/m2, respectively (13). Recurrent coronary outcome events included cardiac death, MI, or unstable angina (hospitalization during follow-up with an increase in either frequency or duration of angina symptoms, or development of new angina at rest—whichever occurred first with both requiring ischemic ECG changes without enzyme elevation). Average patient follow-up was 26 months.
Blood markers
Two months after index MI, fasting blood was drawn, and sera and plasma were prepared. apolipoprotein B (ApoB), total cholesterol, lipoprotein-associated phospholipase A2 (Lp-PLA2), apolipoprotein A-I (ApoA-I), high-density lipoprotein (HDL), triglyceride, low-density lipoprotein (LDL) peak particle diameter, glucose, insulin, plasminogen activator inhibitor-1 (PAI-1), C-reactive protein, von Willebrand factor antigen, fibrinogen, d-dimer, factor VII, and factor VIIa were analyzed as described previously (12,14). Lipoprotein(a) (Lp(a)) was analyzed as described previously (12) using enzyme-linked immunoassay (Macra; Strategic Diagnostics, Newark, DE). Gradient gel electrophoresis was used to fractionate HDL particles according to size (15), the results of which were used to determine HDL median diameter (defined as diameter where half the HDL absorbance is on larger and half on smaller particles).
Genotyping
Genotyping of 17 candidate polymorphisms was performed as described previously (16). Candidate polymorphisms were chosen so that, when dichotomized, the lower prevalent form accounted for at least 25% of patients. Genotyping results were available for ∼88% of patients of the study population.
Statistical analyses
Statistical analyses were performed using Statistica 7.0 (StatSoft, Tulsa, OK). Linear regression was used to adjust variables that were significantly associated with age. Significant differences between groups were assessed using the Mann-Whitney U-test. Significant differences among groups were assessed using the Kruskal-Wallis test with post hoc testing using Mann-Whitney U-test with Bonferroni correction. Comparison of proportions and genotypic distributions were performed using the χ2-test. Significance level for all analyses was P < 0.05 unless stated otherwise.
Cox multivariable proportional hazards regressions (blood markers treated as continuous variables; genetic polymorphism treated as binary variables) and Kaplan-Meier analysis with log-rank statistics were used to follow outcomes over time. Cox analyses were performed by initial univariate evaluation of 17 blood and 17 genetic markers with a significance level of P < 0.0015 (0.05/34) reflecting Bonferroni correction for multiple comparisons. Also, the statistical significance (P < 0.1) of clinical covariates (sex, race, smoking, prior MI—any MI occurring before the MI of study enrollment, pulmonary congestion, ejection fraction, and claudication) was determined in univariate analyses. Multivariable models were then formulated for main effects as simultaneous entry of all univariate significant clinical covariates, and blood and genetic marker variables. Medication effects (statins, β blockers, aspirin, calcium channel blockers, nitrates, angiotensin-converting enzyme-inhibitors, and oral anticoagulants; P < 0.05) were assessed by individual addition to multivariable models. Interaction of significant blood marker variables with genetic markers was assessed by inclusion of blood marker/genetic marker interaction terms individually into multivariable models.
Results
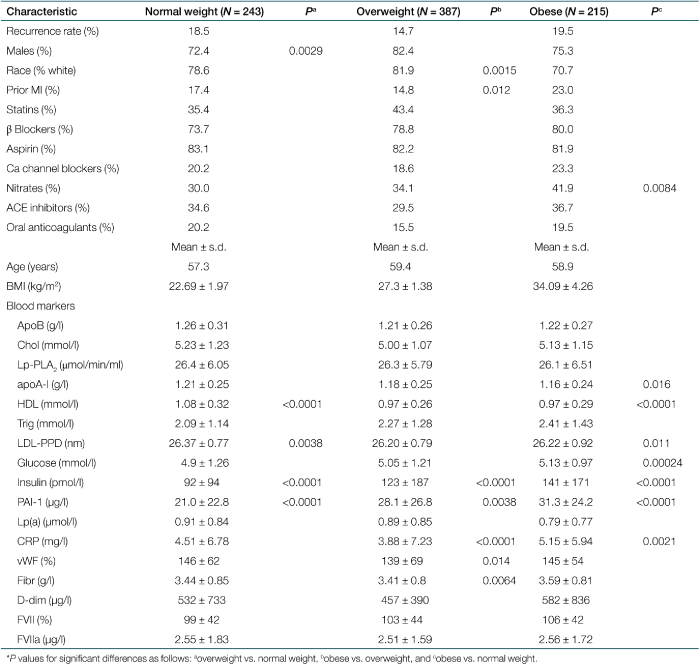
Study population
Recurrent coronary event rates according to outcome events for the three BMI groups were as follows: normal weight −3.7% MI, 2.1% cardiac death, 12.8% angina; overweight −4.4% MI, 1.6% cardiac death, 8.8% angina; and obese −3.7% MI, 1.4% cardiac death, 14.4% angina. Clinical and laboratory characterization of the study population (N = 845) for normal-weight, overweight, and obese patients is given in Table 1. The table shows that recurrent coronary event rates did not differ among the three weight groups, a finding that was confirmed by univariate Cox regression analysis. Table 1 demonstrates statistically significant differences in clinical and laboratory parameters as follows: overweight in comparison to normal weight—more males, higher levels of insulin and PAI-1, lower levels of HDL, and smaller LDL particles; obese in comparison to normal weight—more patients on nitrates, higher levels of glucose, insulin, PAI-1 and C-reactive protein, lower levels of apoA-I and HDL, and smaller LDL particles and obese in comparison to overweight—more black patients, more patients with prior MI, and higher levels of insulin, PAI-1, C-reactive protein, von Willebrand factor, and fibrinogen.
 |
Multivariable models in normal-weight and overweight postinfarction patients
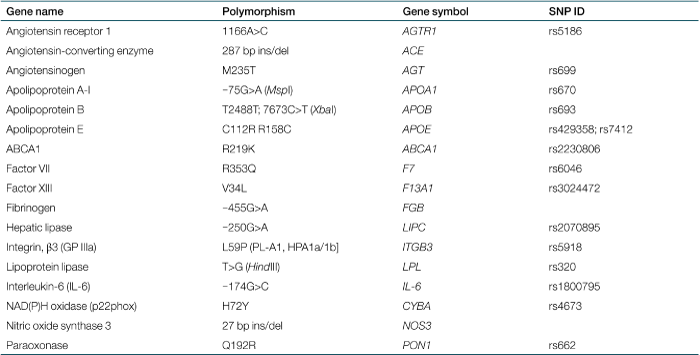
Cox proportional hazards multivariable modeling adjusted for significant clinical covariates was performed separately in normal-weight, overweight, and obese patients with time to recurrence as a function of 17 blood markers (Table 1) and 17 genetic markers (Table 2). Genetic polymorphism were in Hardy-Weinberg equilibrium except those relating to angiotensinogen, hepatic lipase, IL-6, NAD(P)H oxidase, and paraoxonase. Also, in each case, the less frequent variant of dichotomized polymorphisms accounted for at least 25% of the study population.
 |
Results revealed no univariate significant blood markers or polymorphisms for normal-weight or overweight patients that predicted recurrence.
Multivariable models in obese postinfarction patients
Corresponding analyses in obese patients revealed only Lp(a) of the blood markers as a significant univariate factor while no polymorphism was found to be a significant factor. Of clinical covariates only prior MI achieved significance. Multivariable modeling including Lp(a) as a continuous variable and prior MI as a covariate revealed hazard ratio with 95% confidence interval and P value as follows: prior MI −1.91 (1.02–3.59) P = 0.045 and Lp(a) −1.67 (1.26–2.22) P = 0.00039. Individual entry into the model of medication use was performed for statins, β blockers, aspirin, calcium channel blockers, nitrates, angiotensin-converting enzyme inhibitors, and oral anticoagulants. None achieved statistical significance. Kaplan-Meier curves showing recurrent coronary events over time for Lp(a) (dichotomized as upper tertile vs. lower two tertiles, Lp(a) cutpoint was 24 mg/dl) is given in 1. This dichotomization scheme was chosen based on outcome event rates as follows: tertile 1–9.7%, tertile 2–12.5%, and tertile 3–37.7% indicating a robust increase in outcome rate for tertile 3 relative to the other two. To facilitate interpretation of Lp(a) hazard ratio, corresponding Cox multivariable modeling using Lp(a) dichotomized as above was performed to give 3.94 (2.11–7.35) P = 0.000017.

Kaplan-Meier curves for obese postinfarction patients showing recurrent coronary events over time according to Lp(a) concentration (solid line—lower two tertiles of Lp(a); dotted line—upper Lp(a) tertile, P = 0.00001).
Even though race was not a significant clinical covariate in obese patients, additional assessment was performed to further evaluate potential effects of race on results. Forcing race into the multivariable model left results for Lp(a) essentially unchanged as did inclusion of the interaction term of race with Lp(a). Separate Cox multivariable models according to race revealed Lp(a)-associated risk essentially the same in white patients as in black patients although the regression in black patients achieved only borderline significance (P = 0.053) most likely as a result of low patient number (N = 60).
Genotypic interactions of Lp(a) in obese postinfarction patients
To assess potential interaction between Lp(a) and genetic markers (Table 2), Cox multivariable model for obese patients was run with individual addition of an interaction term of Lp(a) with each marker. Results revealed significant interactions of Lp(a) with the −75G>A polymorphism of APOAI and the −250G>A polymorphism of LIPC. The nature of the interactions is demonstrated in 2 by Kaplan-Meier curves corresponding to each dichotomized polymorphism for patients in the upper Lp(a) tertile vs. those in the lower two tertiles of Lp(a). For variants of each polymorphism, patients with high Lp(a) levels were at higher risk; for patients with low Lp(a) levels neither polymorphism was associated with risk.

Kaplan-Meier analyses in obese postinfarction patients demonstrating risk-associated interaction of high Lp(a) with the −75G>A polymorphism of apoA-I and with the −250G>A polymorphism of hepatic lipase. (a) apoA-I polymorphism. For patients with low Lp(a) (lower two tertiles), the curves show no significant difference for patients with the A allele (dotted line, N = 28) vs. patients homozygous for the G allele (solid line, N = 103) (P = 0.43) while for patients with high Lp(a) (upper tertile), the curves are significantly different for patients with the A allele (dotted line, N = 18) vs. patients homozygous for the G allele (solid line, N = 40) (P = 0.013). (b) Hepatic lipase polymorphism. For patients with low Lp(a), the curves show no significant difference for patients homozygous for the G allele (dotted line, N = 79) vs. patients with the A allele (solid line, N = 49) (P = 0.98) while for patients with high Lp(a), the curves trend toward significant difference for patients homozygous for the G allele (dotted line, N = 30) vs. patients with the A allele (solid line, N = 26) (P = 0.10).
HDL and Lp(a)-associated risk
From the previous, recurrent risk in obese patients was found to be associated with interactions of high levels of Lp(a) with polymorphisms in apoA-I and hepatic lipase genes both of which are associated with lipoprotein metabolism, particularly that of HDL. Therefore, we explored further the nature of the above interactions by investigating associations of several HDL measures (HDL cholesterol and apoA-I levels and median HDL particle diameter) with recurrent events in the two groups as defined by Lp(a) levels. HDL cholesterol and apoA-I demonstrated no differences in levels as a function of recurrence for either low or high Lp(a) patients. Median HDL particle diameter demonstrated no difference in size as a function of recurrence in patients with low Lp(a); however, patients with high Lp(a) levels and recurrent events had smaller median HDL diameter in comparison to patients without recurrent events (8.74 ± 0.39 nm vs. 8.81 ± 0.26 nm, P = 0.035).
Discussion
Results of multivariable analysis adjusted for significant clinical covariates in obese postinfarction patients showed only elevated Lp(a) to be an independent risk marker for recurrent coronary events out of a set of 17 thrombogenic, inflammatory, and metabolic blood markers and a set of 17 single-nucleotide polymorphisms of proteins associated with CVD risk. By contrast, none of the above markers were found to be associated with recurrent risk in either normal-weight or overweight postinfarction patients. Further, for obese postinfarction patients, single-nucleotide polymorphism variants of APOAI (−75G>A) and LIPC (−50G>A) were found to interact with elevated Lp(a) in risk association; lastly, for obese postinfarction patients with high Lp(a) levels, HDL particles were smaller in patients with vs. without recurrent events.
Lp(a) is a lipoprotein resembling LDL in lipid makeup, and like LDL, contains one molecule of apoB-100; however, in Lp(a), apoB-100 is attached via disulfide linkage to apolipoprotein(a), a highly glycosylated protein containing a variable number of repeat domains homologous to the kringle IV structure of plasminogen (17,18,19). Lp(a) is thought to be an atherogenic lipoprotein with most, but not all, population studies demonstrating independent association of high Lp(a) levels with CVD risk (17,18,19) including a recent meta-analysis of prospective studies (20). As in the current work on patients with established CVD, several other studies have also demonstrated elevated Lp(a) as associated with risk of recurrent coronary events (21,22); however, adiposity was not considered in these studies. Surprisingly, we have found only one study (23) reporting Lp(a)-associated CVD risk in overweight patients (BMI ≥ 27 kg/m2); but the study involved primary rather than secondary end points.
Several recent Lp(a) studies have focused on the accentuation of Lp(a)-associated CVD risk by its interaction with conventional and emerging risk factors (18). We investigated interactions of Lp(a) with a set of CVD-associated genetic polymorphisms and demonstrated significant risk associations with promoter-region polymorphisms in APOAI (−75G>A) and LIPC (−250G>A), the structural genes for apoA-I and hepatic lipase, respectively. Regarding polymorphism-associated phenotypic effects, there have been inconsistent results reported for the influence of the apoA-I polymorphism on apoA-I and HDL levels (24,25), while for the hepatic lipase polymorphism, increased enzyme activity has been reported (26). Regarding polymorphism-associated CVD risk, from the few studies available for the apoA-I polymorphism, the A allele has been reported to be associated with CVD risk (25,27), which is consistent with our results, whereas for the hepatic lipase polymorphism, results have been mixed (26,28). Results of the current study also demonstrated smaller HDL particles in obese patients with high levels of Lp(a) and recurrence vs. corresponding patients without recurrence while HDL and apoA-I levels were not different as a function of recurrence or Lp(a). Thus, although increased atherogenicity of Lp(a) has been reported in association with low levels of HDL (29,30,31), it was not apparent in the present work; rather, observed findings suggested association of Lp(a) atherogenicity with small HDL particle size. This is consistent with previous reports (32,33) showing that conditions (like obesity) associated with small dense HDL and LDL particles are also associated with small dense (presumably atherogenic) Lp(a) particles. Thus, it is clear that multiple individual associations demonstrated in the current study between obesity, Lp(a), small HDL, and the genetic markers of apoA-I and hepatic lipase interact in a complex fashion to bring about the observed patterns of risk distribution. Mechanistic details underlying the establishment of such risk would require specifically focused studies.
The findings and extent of conclusions of the current study were limited by several factors. Use of BMI as a measure of adiposity was an issue of concern in that BMI does not take into account the distribution of adipose tissue within the body, vis-à-vis, visceral vs. subcutaneous deposition. However, BMI has been a standard measure of adiposity in numerous studies involving obesity and, therefore, serves as a basis for comparison with previous work. Failure to identify blood or genetic risk markers in normal-weight and overweight patients and failure to identify additional risk factors in obese patients may relate to statistically underpowered patient subgroups. On the other hand, the high level of significance of findings of the current study suggests they are robust, although the usual caveats associated with the results of subgroup analyses especially with a large number of independent variables remain as relevant. Additionally, it should be noted that all blood marker levels were determined 2 months postindex MI, which may have particular implications for Lp(a), as elevations in Lp(a) levels extending beyond this period have been reported in postinfarction patients. Such elevations have been thought to be possibly related to Lp(a) acting as an acute-phase reactant. Thus, it may be that the Lp(a) levels of the current study were not baseline values and the observed risk association of Lp(a) may be related to a potential association with the acute-phase response. Also potentially contributing to study limitations was the lack of additional information including especially apo(a) isoforms and clinical covariates potentially related to risk such as development of diabetes, exercise, diet, ethanol use, affective mental status, and social support. Specifically focused future studies would be necessary to resolve theses issues. Strengths of the current work include a prospective study with long-term follow-up along with availability of a set of 17 thrombogenic, inflammatory, and metabolic blood markers, and 17 genetic CVD-associated polymorphisms that were initially screened using Bonferroni correction, P < 0.0015 (0.05/34), to minimize false positives.
In conclusion, results of the current study show for obese postinfarction patients, in comparison to overweight and normal-weight postinfarction patients, a different pattern of risk associations with blood and genetic markers. In obese postinfarction patients, elevated Lp(a) was the only independent marker of risk out of a set of 17 blood and 17 genetic markers in contrast to overweight and normal-weight patients in whom none of the markers was associated with risk. Further, in obese postinfarction patients with elevated Lp(a), interactions with polymorphism variants in APOAI (−75G>A) and LIPC (−250G>A) contributed to risk, and, additionally. in obese patients with high levels of Lp(a) and recurrence, HDL particles were smaller in comparison to obese patients with high Lp(a) levels without recurrence. Findings of the current study in obese postinfarction patients suggest a pattern of altered lipoprotein metabolism, especially as related to HDL, in contributing to establishment of risk for recurrent coronary events in these patients. Given the limitations of the current study regarding results of subgroup analyses with a large number of variables as noted above, findings of the current study need to be replicated in similar populations and investigated in additional study groups to define potential clinical import including potential diminution of risk in these patients with weight reduction.
Acknowledgment
This study was supported by research grant HL-48259 from the National Institutes of Health (NIH), Bethesda, MD, and by a contract from Millennium Pharmaceuticals, Inc., Cambridge, MA. We are indebted to the Study Coordinators who enrolled and followed up the patients from the 13 participating centers. Also, the project described was (in part) supported by Grant Number KL2 RR 024136 from the National Center for Research Resources (NCRR), a component of the NIH and the NIH Roadmap for Medical Research, and its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. Information on NCRR is available at http:www.ncrr.nih.gov. Information on Re-engineering the Clinical Research Enterprise can be obtained from http:nihroadmap.nih.govclinicalresearchoverview-translational.asp.
Disclosure
The authors declared no conflict of interest.

