

Δ98Δ, a minimalist model of antiparallel β-sheet proteins based on intestinal fatty acid binding protein
Abstract
The design of β-barrels has always been a formidable challenge for de novo protein design. For instance, a persistent problem is posed by the intrinsic tendency to associate given by free edges. From the opposite standpoint provided by the redesign of natural motifs, we believe that the intestinal fatty acid binding protein (IFABP) framework allows room for intervention, giving rise to abridged forms from which lessons on β-barrel architecture and stability could be learned. In this context, Δ98Δ (encompassing residues 29–126 of IFABP) emerges as a monomeric variant that folds properly, retaining functional activity, despite lacking extensive stretches involved in the closure of the β-barrel. Spectroscopic probes (fluorescence and circular dichroism) support the existence of a form preserving the essential determinants of the parent structure, albeit endowed with enhanced flexibility. Chemical and physical perturbants reveal cooperative unfolding transitions, with evidence of significant population of intermediate species in equilibrium, structurally akin to those transiently observed in IFABP. The recognition by the natural ligand oleic acid exerts a mild stabilizing effect, being of a greater magnitude than that found for IFABP. In summary, Δ98Δ adopts a monomeric state with a compact core and a loose periphery, thus pointing to the nonintuitive notion that the integrity of the β-barrel can indeed be compromised with no consequence on the ability to attain a native-like and functional fold.
Introduction
Despite the low sequence identity displayed by the members of the fatty acid binding protein (FABP) family, FABPs share a similar β-barrel fold that resembles a clamshell. In particular, intestinal FABP (IFABP) is a monomeric 131 amino acids polypeptide (15 kDa), devoid of C or P residues, thereby avoiding two major problems that complicate the analysis of protein folding.1Because of this fact, IFABP becomes an attractive model to study the structure and dynamics of β-sheet proteins. Briefly, this β-barrel protein consists of two five-stranded β-sheets (βA-βE and βF-βJ) arranged in a nearly orthogonal orientation, enclosing the ligand binding cavity. All β-strands are connected by β-turns with the exception of strands βA and βB, where an intervening helix-turn-helix motif appears.2 Previous work of our laboratory has identified by limited proteolysis an abridged variant of IFABP.3 This fragment (Δ98Δ), although lacking one-quarter of the sequence of the parent protein (it includes only 98 amino acids corresponding to sequence 29–126 of IFABP), is a monomeric stable truncated form of IFABP. In comparison with the full-length protein, Δ98Δ (11 kDa) is devoid of β-strand A, most of the helical domain and the last five amino acids belonging to the C-terminal β-strand (see Fig. 1). Most significantly, this truncation leads to the loss of both stretches involved in the closure of the β-barrel. In IFABP this region comprises a hydrogen bonding network involving residues of the distal part of strands βA and βJ, each of them belonging to different β-sheets. Twisting of strand βA allows contact with its neighbors: the N-terminal end contacts βB, whereas the C-terminal end interacts with βJ. Despite this fact, cumulative evidence indicates that this fragment retains substantial β-sheet content and native tertiary interactions. The reasons for this remarkable behavior would lie on the ancillary role played by the segments deleted and on the conservation of all the critical residues of the hydrophobic core, that is, those involved in the nucleation event leading to the folded state. According to the hierarchical folding mechanism proposed for IFABP,4-6 the unfolded polypeptide first collapses into a semicompact structure around a hydrophobic core composed by F47, F62, L64, F68, W82, M84, and L89 (see Fig. 1). Next, strands βB-βG propagate outwardly from the hydrophobic core, establishing the native topology. Finally, this early structure serves as a scaffold to fold the last three strands (βH–βJ), thus consolidating the native hydrogen bonding network. In agreement with this model, in principle, Δ98Δ would be able to complete most of the stages that lead to the folded state. This abridged variant is the smallest structure of its kind described so far preserving binding activity.

Ribbon structure of IFABP (PDB 2IFB) where the Δ98Δ construct is indicated. The excised N- and C-termini are shown in blue, Δ98Δ in light blue, and residues belonging to the hydrophobic core are depicted with their side chains in CPK representation (in pink). The only W present in Δ98Δ is shown in red.
In this sense, to define the minimal sequence compatible with a stable fold and binding activity is an issue with general implications for protein folding and function. Here we present a comprehensive biophysical characterization of Δ98Δ that fully accounts for its structural and functional features: the conservation of a compact hydrophobic core and the presence of an expanded loose periphery, thereby preserving the ability to interact with amphipathic ligands. Ultimately, we postulate that lessons learnt from this structure might shed light on equilibrium folding intermediates present in the full-length protein.
Results
The monomeric state of the truncated protein was assessed by static light-scattering. When sampled onto a size exclusion chromatography column (Superdex-200), the elution peak obtained for this abridged variant corresponds to a monodisperse protein of 10,260 ± 2565 Da.
Quenching of intrinsic fluorescence intensity
W82, which is buried in the hydrophobic core, is the main contributor to IFABP. Proof of this is the fact that a mutant IFABP lacking W6 yields ∼65% of the fluorescence intensity of the wild type protein.7 Significantly, in Δ98Δ removal of W6 might not suffice to explain the observed decrease in fluorescence intensity. However, the conservation of the center of mass of fluorescence emission between the fragment and the parent protein points to the preservation of the integrity of a common hydrophobic core.3 In this regard, to evaluate the solvent accessibility of the core, we measured the effect of two quenchers on protein fluorescence. The anion iodide (KI) is an effective quencher for both proteins [Fig. 2(A)], the abridged variant being more affected than the parent protein: Stern-Volmer constants (Ksv): 3.80 ± 0.22 and 1.52 ± 0.09 M−1, respectively. In addition, quenching by the neutral molecule acrylamide was investigated at five different temperatures, ranging from 21.3 to 35.0°C [Fig. 2(B)]. Here again, Ksv values for Δ98Δ (i) are consistently higher and (ii) show a more marked temperature dependence than those for IFABP. The latter evidence points to a collisional quenching process. It is noteworthy that the addition of the fatty acid ligand exerts a stronger effect on the fluorescence quenching of Δ98Δ, which manifests itself as lower Ksv values and a less apparent dependence on temperature.

Quenching of intrinsic fluorescence intensity. (A) Iodide quenching of apo-IFABP (□) and apo-Δ98Δ (○). (B) Arrhenius plot for the acrylamide quenching of apo-Δ98Δ (○), holo-Δ98Δ (•), apo-IFABP (□), and holo-IFABP (▪).
Equilibrium unfolding studies
Either by measuring the change of the center of mass of fluorescence spectra or the dichroic signal at 216 nm, the temperature-induced unfolding of the abridged variant shows a cooperative behavior (see Fig. 3). As far as the issue of reversibility is concerned, Δ98Δ follows a cooling curve superimposable to the heating data (after heating up to ∼81°C, a temperature well above the midpoint of the transition; data not shown). The transition shown by Δ98Δ is less cooperative than that observed for IFABP, the former exhibiting a lower midpoint temperature of denaturation (Tm): 71 and 79°C, for the truncated and wild-type proteins, respectively [Fig. 3(A)]. In each case, the Tm values measured for the holo-forms are indistinguishable from those of the apo-forms. These data suggest that near the Tm hardly any fatty acid remains bound to the protein. This is consistent with the fact that binding activity of IFABP assayed by the Lipidex method sharply diminishes as temperature rises, being almost nil at 70°C.8 On the other hand, the center of mass of IFABP remains constant up to ∼70°C, while the pretransition for the abridged variant shows a marked positive slope.

Thermally induced equilibrium unfolding transitions of apo-Δ98Δ (○), holo-Δ98Δ (•), apo-IFABP (□), and holo-IFABP (▪). The transitions were monitored by the change in the center of mass of the fluorescence emission spectra (A), and by the evolution of the molar ellipticity at 216 nm (B), where the left and right ordinate axes correspond to IFABP and Δ98Δ, respectively.
Similar features are observed for the curves describing the loss of secondary structure as a function of temperature [Fig. 3(B)]. Δ98Δ behaves less cooperatively than IFABP, showing a pretransition region of significant positive slope, partially masking the smaller amplitude transition region. These facts make the calculation of Tm for the abridged variant less certain than that for the full-length protein (Tm: 61 and 77°C for the truncated and wild-type proteins, respectively), pointing to the fact that Δ98Δ is indeed able to attain a folded form, albeit displaying increased conformational flexibility. Here again, upon oleic acid addition, no significant stabilization occurs, as judged by the invariance of the Tm values.
The larger difference in Tm observed between circular dichroism and fluorescence data sets corresponding to Δ98Δ uncovers the presence of partially folded species populated in this variant. At variance with most structures of this kind reported for other proteins, the above forms display a reduced content of secondary structure but would preserve as well a fairly conserved environment around W82 (see “Discussion” section). In this sense, stopped-flow fluorescence kinetic experiments following the fluorescence signal have shown that at least one intermediate species would be present on each path along the unfolding and refolding of IFABP.1, 9-11 Suggestively, this form shows little if any secondary structure but maintains some tertiary contacts. The latter interactions would be the first to form during folding and the last to break down during unfolding.11
Figure 4 shows the GdnHCl-induced equilibrium unfolding transition for IFABP and Δ98Δ measured by circular dichroism and fluorescence spectroscopies. All data describe cooperative transitions that, considered individually, seemingly reflect two-state processes. Nevertheless, upon closer examination, the fraction of native state plotted in Figure 5 clearly points to a diverse scenario, where Δ98Δ exhibits the most complex behavior. Consistently, Δ98Δ (i) unfolds cooperatively, (ii) is less stable than the parent protein, and (iii) is stabilized to a larger extent than IFABP by the ligand fatty acid (see Cm values in Table I). The latter finds a counterpart in limited proteolysis experiments (see below). The intensity of the fluorescence emission clearly changes at lower denaturant concentrations than the secondary structure probe (far-UV CD). This can be interpreted as a proof that for Δ98Δ significant amounts of intermediates—showing substantially larger solvent accessibility—are being populated at equilibrium. Nevertheless, it is revealing to point out that the center of mass of fluorescence emission [CMFE; see Fig. 4(D)] varies along the transition resembling more closely but still preceding the pattern shown by the CD data. CMFE data were not processed further, because this parameter and also the maximum wavelength of fluorescence emission (λmax)12 can be proven in general to hold a nonlinear relationship with the molar fraction of native or unfolded protein (results not shown). On the other hand, in both apo- and holo-forms of Δ98Δ, conformational changes as monitored by near and far-UV CD lie close together. Thus, from the bulk of this evidence, it is not unreasonable to propose that Δ98Δ might preserve a small cooperative nucleus, showing enhanced solvent exposure but still able to support a β-barrel scaffold.

GdnHCl-induced equilibrium unfolding transitions. Evolution of the ellipticity signal at 216 nm (A), the integrated near-UV CD signal (B), the fluorescence intensity at 330 nm (C), and the center of mass of the intrinsic fluorescence emission (D) as a function of denaturant concentration of apo-Δ98Δ (○), holo-Δ98Δ (•), apo-IFABP (□), and holo-IFABP (▪).

Molar fraction of native form derived from the GdnHCl-induced equilibrium unfolding transitions of apo-Δ98Δ (A), holo-Δ98Δ (B), apo-IFABP (C), and holo-IFABP (D). Transitions were monitored by far-UV (▴) and near-UV CD (▾), and the intensity of fluorescence emission at 330 nm ( ).
).
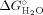 (kcal mol−1)
(kcal mol−1) |
mN-U (kcal mol−1 M−1) | Cm (M) | |
|---|---|---|---|
| IF330nm | |||
| apo-Δ98Δ | 2.33 ± 0.29 | 2.56 ± 0.25 | 0.91 ± 0.03 |
| holo-Δ98Δ | 2.31 ± 0.25 | 2.33 ± 0.16 | 0.99 ± 0.04 |
| apo-IFABP | 8.82 ± 0.84 | 5.65 ± 0.50 | 1.56 ± 0.01 |
| holo-IFABP | 9.28 ± 0.52 | 5.94 ± 0.01 | 1.56 ± 0.01 |
| θ216nm (° cm2 dmol−1) | |||
| apo-Δ98Δ | 3.27 ± 1.29 | 2.95 ± 0.93 | 1.11 ± 0.09 |
| holo-Δ98Δ | 4.60 ± 3.09 | 3.41 ± 2.00 | 1.35 ± 0.12 |
| apo-IFABP | 5.61 ± 0.56 | 3.59 ± 0.29 | 1.56 ± 0.03 |
| holo-IFABP | 6.25 ± 0.63 | 3.75 ± 0.29 | 1.67 ± 0.04 |
| θ integrated in the range 250–320 nm (° cm2 dmol−1) | |||
| apo-Δ98Δ | 3.64 ± 1.38 | 2.92 ± 0.85 | 1.24 ± 0.11 |
| holo-Δ98Δ | 5.41 ± 0.78 | 3.85 ± 0.29 | 1.41 ± 0.01 |
| apo-IFABP | 8.41 ± 0.68 | 5.51 ± 0.29 | 1.53 ± 0.04 |
| holo-IFABP | 15.00 ± 0.63 | 9.62 ± 0.29 | 1.56 ± 0.02 |
- Values are expressed as the means ± standard deviations.
Limited proteolysis
Partial proteolysis can shed light on the location of ligand binding sites or conformational changes in proteins, because this technique reveals the differential exposure of spatially discrete sites along the polypeptide sequence. In the early stages of this work, Arighi et al.13 investigated the peptide pattern arising from IFABP after digestion with clostripain (Arg-C) both in the absence and in the presence of oleic acid. It was found that apo-IFABP was much more sensitive towards protease digestion than holo-IFABP, a fact consistent with a more rigid conformation. In fact, under the conditions then used [20 mM Tris-HCl (pH 7.8), 50 mM NaCl at 37°C], the Δ98Δ fragment prevails in the mixture but smaller peptides were also found to be present. As we have already seen, the addition of oleic acid to the fragment exerts some stabilizing effect upon GdnHCl-induced unfolding (see previous section).
To take further advantage of the protease as a tool to explore conformation, we challenged apo- and holo-Δ98Δ with this enzyme. Both apo-proteins were proteolyzed more extensively than the corresponding forms loaded with the fatty acid, a behavior compatible with a ligand-induced decrease in conformational flexibility (see Fig. 6). Remarkably, under the conditions used in this work (20 mM Tris-HCl, pH 8.0, in the absence of NaCl at 30°C; see “Materials and Methods” section for more details) proteolysis of holo-Δ98Δ generates a ∼9-kDa fragment that is resistant to degradation. The mass of this peptide measured by MALDI-TOF spectrometry is 8809.4 Da, corresponding to the segment 29–106 of IFABP (after taking into account the presence of an N-terminal methionine). Encompassing a 78 amino acids long internal fragment of the parent protein, this new variant was named Δ78Δ. It is noteworthy to underscore that this variant conserves all the structurally relevant amino acids belonging to the hydrophobic core, suggesting that the protease was able to discriminate between flexible and well packed regions. Analogously to Δ98Δ, we propose that the stability towards proteolysis of this fragment is due to its ability to bind the ligand, a feature that would help it to remain structured. This yet smaller abridged variant was cloned, expressed, and purified and is currently being structurally characterized in our laboratory.

Separation by SDS-PAGE of the digestion mixture of Δ98Δ and IFABP after an overnight treatment with clostripain. Proteolysis was carried out in the presence (holo) or in the absence (apo) of oleic acid, as indicated in the “Materials and Methods” section.
Probing the conformation of Δ98Δ with fluorescent ligands
Binding of the fluorescent probe 1-anilino naphthalene-8-sulfonic acid
1-Anilino naphthalene-8-sulfonic acid (ANS) is a fluorescent probe widely used for the diagnosis of molten globule states. Unlike most native proteins, IFABP naturally binds ANS with a stoichiometry of 1:1.13-15 This complex could be disrupted by the natural ligand oleic acid, indicating that the bound probe localizes within the binding cavity of IFABP.13 In this context, it is not unreasonable to speculate that (i) the sulfonic group of ANS could mimic the carboxylate group, and (ii) the nonpolar aromatic moiety might effectively resemble the alkyl chain of the fatty acid.8, 15 The X-ray crystallographic structure of IFABP bound to palmitate shows that the fatty acid carboxylate indeed interacts with the guanidinium moiety of R106 in the binding pocket,16 therefore ANS would be sensing a region located deeply inside the binding cavity. Further structural insights arose from comparative measurements of ANS binding activity of the truncated variant and IFABP. For each protein, Figure 7(A) depicts the evolution of the fluorescence intensity as a function of the concentration of the fluorophore. Initially, nonlinear regression to a model of n identical and noninteracting binding sites was fitted to the data. Results indicate that the full-length protein would present a single binding site (n = 0.7 ± 0.1, Kd = 23 ± 3 μM), whereas this analysis applied to Δ98Δ yields less certain values (n = 1.2 ± 0.1, Kd = 133 ± 8 μM), because of (i) the lesser affinity shown by the variant and (ii) the limited range of ligand concentration attainable in the assay. Thus, the existence of a second binding site cannot be ruled out by this data (see below).

Binding of ANS to Δ98Δ and IFABP. (A) Titration of Δ98Δ (○) and IFABP (□) with the fluorescent probe. (B) Competition of ANS bound to Δ98Δ (○) and IFABP (□) by oleic acid. The continuous lines correspond to the fitting of Eq. (5) (see “Materials and Methods” section) to the data. The values of the parameters thus derived are the following: ΔF0 = 0.67, Kdapp = 3.8 μM and ΔFres = 0.33 for Δ98Δ; and ΔF0 = 0.98, Kdapp = 0.14 μM, ΔFres = 0.02 for IFABP.
The analysis of the emission spectrum of ANS bound to each of the proteins sheds light on the nature of the binding site (result not shown). The position of the maximum of emission (λmax) obtained for the IFABP-ANS complex (479 nm) falls beyond the red extreme of the range (about 468–477 nm) usually reported for ANS-protein complexes and is in agreement with previously published values.13, 14 This fact reveals that the natural binding pocket present in IFABP represents a hydrophilic environment, quite unlike the case of typical molten globules. Conversely, for the ANS-Δ98Δ complex, the measured λmax (466 nm) is similar to those reported for apomyoglobin and albumin complexed to ANS (454 and 465 nm, respectively14). In addition, the ANS-Δ98Δ complex results in enhanced fluorescence intensity as compared to IFABP. This evidence put together—the increased quantum yield and the shift to a lower wavelength of the maximum of emission—could be interpreted by assuming that the binding site(s) for ANS are more hydrophobic in nature than that present in IFABP.
Competition assays serve to add further insight into this matter [Fig. 7(B)]. Interestingly, while oleic acid completely displaces the ANS bound to IFABP, this is not the case for Δ98Δ, where ∼50% of fluorescence intensity remains even at the highest concentration of competing ligand assayed. A model that contemplates this behavior, that is, one displaceable site plus a nondisplaceable site [Eq. (5)] (see “Materials and Methods” section), yields a ΔFres value of 0.33 and a Kdapp of 3.8 μM. No significant difference exists in the λmax of fluorescence emission between samples assayed in the absence or in the presence of an excess amount of oleic acid, providing confirmatory evidence on the hydrophobic character of the sites probed by ANS (data not shown).
Binding of trans-parinaric acid: circular dichroism analysis
trans-Parinaric acid is a naturally occurring 18-carbon fluorescent polyunsaturated fatty acid (18:4) with advantageous spectroscopic properties that make it a useful biophysical probe.17, 18 The affinity of IFABP and Δ98Δ for trans-parinaric acid was studied through the enhancement of the emission intensity of the bound probe. Interestingly, Δ98Δ retains the ability to bind this fatty acid (Kd = 0.72 μM), although displaying a fivefold lower affinity than IFABP (Kd = 0.13 μM).3trans-Parinaric acid exhibits a strong π→π* transition above 300 nm, a region where most proteins hardly absorb light and, because of its symmetrical nature, this chromophore shows no optical activity either in organic or in aqueous solutions.19 These characteristics enabled its use as a probe to study protein binding sites by circular dichroism.19-21 The circular dichroism spectrum of the complex between Δ98Δ and trans-parinaric acid was compared with that obtained for its apo-form (see Fig. 8). The complex shows a strong negative band at ∼247 and a positive band at ∼238 nm, which in all likelihood arise as a consequence of chromophore binding. For comparison, binding of trans-parinaric acid to IFABP induces two similar bands -albeit of smaller magnitude and opposite sign. In addition, a positive band centered at ∼320 nm and other well-resolved positive bands in the 250–300 nm region appear, that likely represent a mixed contribution of IFABP aromatic residues and induced bands of the probe (results not shown). Being more rigid, the full-length protein would be able to create a more asymmetric environment around the ligand, as evidenced by the presence a fine-structured spectrum. By contrast, an enhanced conformational flexibility of Δ98Δ might explain its dissimilar behavior.

UV circular dichroism spectra of Δ98Δ in the presence (dashed line) or in the absence (continuous line) of trans-parinaric acid. Cell paths of 10 and 1 mm were used for the 250–320 (left axis) and 200–250 (right axis) regions, respectively. The protein:probe molar ratio was 1:0.7.
Discussion
Δ98Δ, a form that results from a major truncation of IFABP, is a very interesting model to study critical features determining folding and function of β-barrel proteins. In this sense, to define the minimal sequence compatible with a stable fold and the preservation of binding activity is an issue with general implications for this class of proteins. Although lacking stretches involved in the closure of the β-barrel, Δ98Δ is stable in solution. In this regard, several truncated variants of IFABP have been reported. One example is IFABP1–128, generated by deleting the C-terminal tripeptide of the parent protein. This variant is described as a monomeric and compact protein, highly susceptible to proteolytic attack and displaying an unfolding behavior indicating a loose tertiary structure.22 In addition, two generations of helix-less variants (Δ17-SG and Δ27-GG)—designed by removing the whole helical domain and replacing it with a dipeptide linker—were found to maintain the same overall topology of the parent protein.23-25 These forms differ from Δ98Δ, in that the latter was uncovered by partial proteolysis of IFABP rather than by rational design. In a more general context, protein truncation has been extensively used to populate nonnative states under physiological conditions.26-28
In this article, we present a thorough biophysical characterization for the construct Δ98Δ that allowed us to postulate a structural model. To this end, we first evaluated the solvent accessibility of the hydrophobic core, after assaying the effect of two quenchers of different chemical nature (iodide and acrylamide) on the fluorescence emission of the single remaining tryptophan in the molecule (W82). Regardless of which quencher was used, the decrease in fluorescence intensity was larger for Δ98Δ than for IFABP, pointing to a greater accessibility to these compounds because of lesser steric restrictions, an expected result considering that Δ98Δ lacks the β-barrel lid. In addition, the abridged variant displays a stronger dependence of Ksv with temperature, suggesting an increased conformational flexibility than the parent protein. It should be pointed out that the range of temperatures employed is far from that where the full-length protein undergoes a structural rearrangement around phenylalanine residues8 and well below the measured denaturation point of both proteins (see Fig. 3). It is noteworthy that the addition of the fatty acid ligand on Δ98Δ gives rise to lower Ksv values and a lesser dependence of this parameter on temperature. Previous work had reported that all tryptophan residues in apo-IFABP are accessible to acrylamide and that bound oleic acid effectively decreases the accessibility of W82 of IFABP, without necessarily implicating any significant structural change of the protein.29 In contrast, the effect exerted by fatty acid binding on Δ98Δ is primarily due to ligand-induced conformational rearrangements, as is supported by data on GdnHCl and temperature-induced conformational transitions and partial proteolysis.
As we have postulated, Δ98Δ would be able to complete at least the first key steps in the folding mechanism proposed for IFABP,4, 6 namely, those involving the hydrophobic collapse and the propagation of strands βB–βG, after which the native topology would be established. For the full-length protein, the last step would involve the folding of strands βH–βJ, thus completing the native hydrogen bonding network. Clearly, Δ98Δ constitutes a folding unit exhibiting significant cooperativity (Figs. 3-5). Nevertheless, this character does not rule out the existence of intermediate species that are indeed revealed both by thermal or chemical (GdnHCl) perturbation. In this regard, the intensity of the fluorescence emission dissociates from the rest of the conformational probes, advancing changes at a lower range of denaturant concentration [Figs. 5(A,B) and 4(D)]. Including the center of mass parameter in this analysis provides a (qualitative) picture uncovering the environment around W82, the key residue reporting on the status of the hydrophobic core. This unusual behavior might find a plausible explanation if the intermediate species, which would preserve a well-folded core, experience increased quenching by the aqueous solvent and/or by amino acid side-chains belonging to the more mobile periphery as unfolding proceeds.
By its very nature, Δ98Δ would constitute itself into a minimalist model potentially capable of populating discrete intermediates sparsely represented in the conformational ensemble of the full-length protein. This goes in line with the idea of constructing a template capable of satisfying hydrophobic interactions prior to β-sheet structure formation as a means of understanding the folding of antiparallel β-sheet proteins.30 In fact, evidence for equilibrium intermediates in IFABP—structurally akin to Δ98Δ—has already been reported by NMR.31, 32
Moreover, despite the extensive truncation, Δ98Δ retains the ability to bind oleic acid. This event would result crucial for the stabilization of side-chain contacts leading to an ultimate readjustment of the tertiary structure. Further proof of such effect is found in the GdnHCl-induced unfolding curves (Figs. 4 and 5), where the ligand fatty acid exerts a more noticeable stabilizing effect on Δ98Δ. A counterpart to this behavior is found in results from limited proteolysis experiments. Similarly to IFABP, the holo-form of Δ98Δ is considerably more resistant than the apo-form and gives rise to a limiting fragment (Δ78Δ; Fig. 6). Clearly, binding of the fatty acid consolidates the structure of the parent variant and prevents it from being further degraded by the protease.
The fluorescent probe ANS sheds light on the structure of Δ98Δ. Although this variant binds the probe less tightly than the parent protein, it still exhibits an affinity well above that shown by a typical molten globule state [Fig. 7(A)]. More insightful is the qualitative picture provided by the comparison of the binding behavior against the natural ligand oleic acid [Fig. 7(B)]. This fatty acid is indeed able to displace a major part of the bound fluorophore ANS, pointing to the preservation of the fatty acid binding site in the variant. In support of this view, R106, a residue that interacts with the carboxylate group of fatty acids and that is proposed to interact with the sulfonate group of ANS,15 lies very close to W82. In turn, the latter belongs to the compact core of the protein, a region that has been proven to suffer no significant conformational modification upon ligand binding. On the other hand, it is important to note that a substantial amount of the fluorescent probe remains bound to the protein, even in the presence of a large excess of oleic acid, revealing the existence of a binding site in the variant that is absent in the parent protein. In addition, it is noteworthy that Δ98Δ binds ANS and the natural ligands oleic acid and trans-parinaric acid, consistently showing fivefold lower affinities than those measured for the full-length protein.3 This fact underpins the argument further in favor of the consequence on binding of the absence of the helix-turn-helix domain in the construct. However, binding of trans-parinaric acid (see Fig. 8) exposes an enhanced flexibility of the fragment by comparison with IFABP. This fact is highlighted by the absence of narrow well-resolved induced bands in the near-UV region of the complex formed with Δ98Δ (as opposed to that formed with IFABP), whereas a large spectral change occurs in the far-UV region (that is less evident in the complex with IFABP).
In a general sense, this work aims in the direction of helping to understand the folding and stability of a paradigmatic β-barrel protein represented by IFABP. With the aid of a natural tool sensitive to local flexibility (the protease) it was possible to deconstruct the β-barrel, thus dissecting a well-known motif. As a result, a functional abridged variant was generated that maintains critical determinants of the parent structure—as represented by a conserved cohesive core region—despite compromising the hydrogen-bonding network. The information presented here leads to the counterintuitive notion that a β-barrel can do without individual strands and still be able to retain structure and function, thus opening the way to further remodeling of this structure. Lessons learnt on the building of β-barrel proteins might bear interesting implications on the understanding of biologically relevant phenomena, such as the avoidance of aggregation,33 the genesis of amyloid fibrils,34 and the possibility of β-intervention in protein–protein interactions.35
Abbreviations: ANS, 1-anilino naphthalene-8-sulfonic acid; apo- or holo-, prefixes that denote the absence or presence of fatty acid ligand, respectively; CMFE, center of mass of fluorescence emission; GdnHCl, guanidinium hydrochloride; IFABP, intestinal fatty acid binding protein; Δ98Δ, a truncated variant of IFABP corresponding to the fragment 29–126 of the parent protein.
Materials and Methods
Materials
Rat IFABP cDNA, coded in the plasmid pET-11a, was expressed in Escherichia coli strain BL21(DE3) and the protein was purified as described previously.13 Recombinant Δ98Δ coded in the plasmid pET-22b(+), was expressed in E. coli strain BL26.3trans-Parinaric acid was supplied by Molecular Probes (Eugene, OR). ANS, oleic acid, guanidinium hydrochloride (GdnHCl), acrylamide, potassium iodide, potassium chloride, sodium thiosulfate, and buffers were purchased from Sigma-Aldrich (St. Louis, MO).
Ligands
In experiments involving holo-proteins, a 10 mM solution of oleic acid in ethanol was added under stirring to the protein dissolved in buffer A (4:1 fatty acid to protein molar ratio) and the mixture was incubated for at least 40 min at 37°C. The final concentration of ethanol in the assay never exceeded 2% (v/v). Oleic acid purity was ascertained by 1H NMR spectroscopy in a Bruker MSL 300 spectrometer. trans-Parinaric acid and ANS concentrations were estimated by ultraviolet absorption in ethanol: ε306 nm = 77,000 M−1 cm−1 and ε372 nm = 7800 M−1 cm−1, respectively. To rule out the photolytic damage to trans-parinaric acid in the course of spectroscopic measurements, the conservation of the ultraviolet spectra was ascertained by comparison of that at the start with that at the end of each experiment. The purity of ANS was checked by thin layer chromatography (TLC) developed in chloroform:methanol:water (65:25:4, by vol). Spots on the TLC plates were detected by their intrinsic fluorescence and by quenching under illumination with a UV source (254 nm). Analysis of the probe showed a single spot with an RF of ∼0.50, which agrees with the results obtained by Muesing and Nishida.36
Purification of Δ98D
Purification of recombinant Δ98Δ was performed as follows: Cells were lysed with lysozyme in the presence of DNAse I.37 The cellular pellet containing the inclusion bodies was isolated by centrifugation (at 14,500 g for 60 min at 4°C), washed with buffer A (20 mM Tris-HCl, pH 8.0) added with MgCl2 and DNAse I at a final concentration of 10 mM and 10 μg mL−1, respectively, and incubated for 15 min at room temperature. After dissolution of the inclusion bodies in buffer A containing 5 mM glycine and 2M urea, the sample was centrifuged (27,000g for 20 min at 4°C) and the supernatant applied to a Sephadex G-100 column (2.7 × 93 cm2) equilibrated and eluted with buffer A. Subsequently, fractions containing Δ98Δ were pooled and sampled onto an anion exchange column (Whatman DE-52, 2.5 × 3 cm2). Elution was carried out in a single step with buffer B (buffer A added with 100 mM NaCl). Finally, pooled fractions containing the protein were dialyzed against buffer A and stored at −20°C in the presence of 10% glycerol. The identity and purity of proteins along the purification was ascertained by Tris-Tricine SDS-PAGE.38 The composition of stacking and running gels was 4% T, 3% C and 16.5% T, 3% C, respectively. An initial voltage of 50 V was applied until the sample reached the running gel, then it was raised to 100 V for the remainder of the run. Gels were stained with 0.1% (w/v) colloidal Coomassie Brilliant Blue G dissolved in 2% (v/v) H3PO4, 15% (w/v) (NH4)2SO4 for 2 h and then destained with distilled water. Digital images were processed with the Gel-Pro Analyzer Software (Media Cybernetics, Silver Spring, MD).
Fluorescence measurements
Steady-state fluorescence measurements were performed in an Aminco Bowman Series 2 spectrofluorometer operating in the ratio mode and equipped with a thermostated cell holder connected to a circulating water bath. The use of a 1-cm path cuvette sealed with a Teflon cap allowed continuous stirring of the sample with a magnetic bar. When the intrinsic fluorescence of proteins was measured, excitation wavelength was 295 nm and emission was collected in the range 310–400 nm. In this case, the spectral slit-widths were set to 4 nm for both monochromators. For ANS, the excitation wavelength was 400 nm and emission spectra were collected in the range 420–600 nm, the spectral slit-widths for each monochromator being 4 and 8 nm, respectively. When necessary, data were corrected for dilution and inner filter effects.39 For each spectrum, either the wavelength of the center of mass,40 the intensity at the maximum of emission or the total integrated intensity were the parameters used for further analysis.
Circular dichroism
Spectra were recorded on a Jasco J-810 spectropolarimeter. Data in the near-UV (250–320 nm) or in the far-UV (200–250 nm) regions were collected using 10- or 1-mm path cuvettes, respectively. A scan speed of 20 nm min−1 with a time constant of 1 s was used. Each spectrum was measured at least three times and the data was averaged to reduce noise. Molar ellipticity was calculated as described elsewhere,41 using mean residue weight values of 114.45 or 112.01 for IFABP or Δ98Δ, respectively. When needed, temperature control was achieved with a Peltier cell equipped with a magnetic stirrer.
Equilibrium unfolding studies
Conformational transitions were monitored as a function of temperature or denaturant concentration by measuring the change in the intrinsic fluorescence intensity and/or the dichroic signal in the far- and near-UV regions. GdnHCl stock solutions were prepared on the same day of the experiment. Individual samples in denaturant concentration ranging from 0 to 3M GdnHCl were obtained by dilution of a fixed volume of a stock solution of protein in mixtures of buffer B and 8M GdnHCl (∼0.25 mg mL−1 final protein concentration). After incubation for at least 1 h to ensure that the equilibrium had been reached, samples were analyzed by circular dichroism following the loss of (i) secondary structure (by the signal at 216 nm) and (ii) tertiary interactions (by the near-UV signal). In the latter case, given the low magnitude and intrinsic noise of signals, and the fact that parallel changes due to unfolding occur over the full spectral range, transitions were pictured by summing the absolute value of signals at different wavelengths so as to maximize the signal-to-noise ratio. Data were corrected for the background signal of buffer and denaturant. Nonlinear least-squares fits to the equilibrium data were achieved using an equation representing a two-state model for protein denaturation adapted from Santoro and Bolem.42
 ()
() ()
()Fluorescence quenching
 ()
()A charged collisional quencher, potassium iodide, was also used to probe the solvent accessibility of the fluorophore. Individual samples were prepared by diluting a stock solution of protein in mixtures of KI and KCl, and measured at 25°C. The final protein concentration was 5 μM and the total salt concentration was kept constant at 0.3M. Stock solutions contained 10 mM sodium thiosulfate to suppress the formation of iodine.
Titration experiments
Binding of ANS to IFABP or Δ98Δ was monitored by changes in (i) the fluorescence intensity of the dye, (ii) the shift of the center of mass, or λmax of the spectra corresponding to the bound probe. Protein concentration was 2 μM in 20 mM potassium phosphates buffer (pH 7.4). The measurements were recorded at 25°C, after equilibration for 3 min.
Reverse titration (at a fixed ligand concentration)
To estimate the enhancement in fluorescence intensity at saturation (ΔFmax), increasing amounts of each protein were added to a ligand solution at an initial concentration of 1 μM. A double reciprocal plot of the total concentration of protein ([P]total) as a function of the enhancement in fluorescence intensity observed (ΔF) allowed us to calculate the extrapolated value at infinite protein concentration (1/[P]total → 0). This value represents the maximum fluorescence intensity attainable by the ligand when it is completely bound to the protein. ΔF is the difference between the observed fluorescence intensity in the presence (F) and in the absence (F0) of protein.
Titration at a constant protein concentration
 ()
()Competition experiments
 ()
()Limited proteolysis
Clostripain (Arg-C; Sigma) was activated by incubation in buffer A added with 1 mM DTT for 2 h prior to its use. Both apo- and holo-IFABP and Δ98Δ (0.25 mg mL−1) dissolved in the same buffer were incubated at 30°C for at least 15 min before protease addition. Digestion was carried out overnight at the same temperature with a mass ratio of protein to protease of 20:1. Finally, the samples were mixed with sample buffer, heated for 3 min at 96°C, and analyzed by Tris-Tricine SDS-PAGE, as previously described.
Acknowledgements
The authors thank Ms. Gabriela Gómez for her critical reading of the final version of this manuscript.

