A unique binding mode of the eukaryotic translation initiation factor 4E for guiding the design of novel peptide inhibitors
Referenced in the special issue in Honor of Ron Levy; PRO. vol. 25 # 1
Abstract
The interaction between the eukaryotic translation initiation factor 4E (eIF4E) and eIF4E binding proteins (4E-BP) is a promising template for the inhibition of eIF4E and the treatment of diseases such as cancer and a spectrum of autism disorders, including the Fragile X syndrome (FXS). Here, we report an atomically detailed model of the complex between eIF4E and a peptide fragment of a 4E-BP, the cytoplasmic Fragile X interacting protein (CYFIP1). This model was generated using computer simulations with enhanced sampling from an alchemical replica exchange approach and validated using long molecular dynamics simulations. 4E-BP proteins act as post-transcriptional regulators by binding to eIF4E and preventing mRNA translation. Dysregulation of eIF4E activity has been linked to cancer, FXS, and autism spectrum disorders. Therefore, the study of the mechanism of inhibition of eIF4E by 4E-BPs is key to the development of drug therapies targeting this regulatory pathways. The results obtained in this work indicate that CYFIP1 interacts with eIF4E by an unique mode not shared by other 4E-BP proteins and elucidate the mechanism by which CYFIP1 interacts with eIF4E despite having a sequence binding motif significantly different from most 4E-BPs. Our study suggests an alternative strategy for the design of eIF4E inhibitor peptides with superior potency and specificity than currently available.
Introduction
The control of mRNA translation is a key step in several cellular processes and must be finely regulated. Molecules influencing mRNA translation in humans are the eIF4E-binding proteins (4E-BPs), which repress protein synthesis by binding to the eukaryotic translation initiation factor 4E (eIF4E). These factors are involved in the control of processes such as development of synaptic plasticity1-3 and repression of cellular proliferation. Consequently, dysregulation of eIF4E has been observed in cancer4-6 and intellectual disabilities.7, 8 eIF4E acts by binding to the m7GTP cap structure on the 5′UTR of the mRNA (Fig. 1). Once this complex is formed, eIF4E binds to the eukaryotic translation initiation factor 4 gamma (eIF4G) in order to recruit the 43S preinitiation complex to the mRNA and form the active translation initiation complex.9 4E-BPs compete with eIF4G for the same site of eIF4E (Fig. 1). 4E-BPs and eIF4G share a common binding sequence; the canonical binding motif YXXXXL
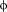 , where Y is tyrosine, X is any residue, L is leucine, and
, where Y is tyrosine, X is any residue, L is leucine, and
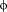 is a hydrophobic residue [Fig. 2(A)].10, 11 It has been shown that this motif forms a conserved α-helix that interacts with eIF4E at the convex site [Fig. 2(B)].11 Phosphorylation of 4E-BPs abolishes their interaction with eIF4E, allowing the positioning of eIF4G in the binding site to promote mRNA translation.12-14
is a hydrophobic residue [Fig. 2(A)].10, 11 It has been shown that this motif forms a conserved α-helix that interacts with eIF4E at the convex site [Fig. 2(B)].11 Phosphorylation of 4E-BPs abolishes their interaction with eIF4E, allowing the positioning of eIF4G in the binding site to promote mRNA translation.12-14

Schematic diagram of the regulatory network of mRNA translation. 4E-BPs (green), eIF4G (light blue), 4E-T (brown), and CYFIP1 (peach) compete for the canonical binding site of eIF4E (blue). Translation of mRNA (gray) is inhibited by CYFIP1, 4E-BP and 4E-T binding. eIF4G is a translational activator.

Summary of the simulation results started from experimental structures of complexes of 4E-BPs with eIF4E. A: Sequence alignment of the 4E-BPs, 4Gs, and CYFIP1 peptides selected for the simulative study. The canonical sequence is reported at the top of the alignment table; the numbering of the residues considered at the binding interface, that is, from 1 to 11, is also shown. The asterisks indicate the residues of the canonical sequence that are not conserved in CYFIP1 (adapted from Ref. 23). B: Superposition of the starting configurations of the 4E-BPs and 4Gs peptides-eIF4E complexes. The side chains of the conserved residues are depicted following the color code reported in panel A. eIF4E is reported in salmon. C: 4E-BPs and 4Gs peptides all-atoms RMSD relative to the starting structure for the trajectory of 1WKW = black, 1EJH = red, 3AM7 = green, 4AZA = yellow, and 1RF8 = violet. D: 4E-BPs and 4Gs peptides helix content calculated as a function of time. Color code as in panel C.
In addition to the canonical site shared with eIF4G, 4E-BPs are also known to interact with eIF4E through a secondary site distinct but proximal to the canonical site. The region of the 4E-BPs involved in this interaction is connected by a linker of 15–30 amino acids to the canonical binding motif.15-17 This secondary binding interaction, not present in eIF4G, has been found to contribute significantly to the affinity of 4E-BPs for eIF4E.18-20
The regulation of eIF4E by 4E-BPs is of significant medical interest. Impairment of the physiological inhibition by 4E-BPs causes unchecked cellular proliferation and has been linked to cancer.4-6, 21 Impairment of eIF4E inhibition has also been linked to autism spectrum disorders (ASDs)1, 8, 22 and specifically to Fragile X Syndrome (FXS).23-26 FXS is caused by the loss or mutation of the fragile X mental retardation protein (FMRP),27 causing upregulation of protein synthesis in brain cells.
One of the mechanisms of translation repression by FMRP involves the control of initiation23, 26, 28, 29 through 4E-BPs. FMRP forms a mRNA translation inhibition complex with the cytoplasmic Fmrp Interacting Protein 1 (CYFIP1).30, 31 CYFIP1, also known as Specific Rac 1 Interacting protein, has been shown to belong to the 4E-BP family,23, 24, 26, 32 and, similarly to other 4E-BPs, it interacts with eIF4E at the canonical binding site23 through a short region (amino acids 721–734), referred in this work as the CYFIP1 peptide (CYFIP1p, for short). However, it does so by means of an unique binding motif.23 In CYFIP1p, the tyrosine in position 1 and the leucine in position 6 characteristic of the canonical eIF4E binding motif are substituted by a leucine and an arginine, respectively, while the hydrophobic residue in position 7 is replaced by the hydrophilic serine [Fig. 2(A)].23 The mode of interaction of CYFIP1 with eIF4E remains an outstanding topic of investigation. The CYFIP1-eIF4E binding motif, with markedly distinct physical characteristics from those of the canonical motif, is therefore expected to display a unique binding mode with eIF4E. The mode of interaction between CYFIP1 and eIF4E can provide additional insights on the fine-tuning of gene expression and possibly on the molecular causes of autism and other disabilities marked by increased protein synthesis.3 This line of research can at the same time provide alternative strategies for the design of inhibitor peptides targeting eIF4E.
Whereas the binding of 4E-BPs and eIF4G to eIF4E has been structurally and biochemically investigated, detailed structural information about the mode of interaction between CYFIP1 and eIF4E is lacking. Biochemical and modeling data have provided evidence for the effects of amino acid mutations in the CYFIP1 eIF4E binding region.23 A structural model of CYFIP1 has been derived from the crystal structure of the wave regulatory complex (WRC),33 a multi-protein complex which regulates cytoskeleton remodeling. A crystallographic model of CYFIP1 bound to eIF4E is not currently available.
In an effort to begin filling this knowledge gap, here, we report the results of atomistic molecular dynamics (MD) simulations aimed at characterizing the dynamics and the likely mode of interaction of the CYFIP1 region responsible for the interaction with eIF4E, and at comparing them to those of other 4E-BPs and eIF4G. Starting with reported crystal structures of 4E-BP- and eIF4G-derived peptides bound to eIF4E and a preliminary model of the CYFIP1-eIF4E binding mode based on biochemical data and protein-protein docking (Tomoo et al., Crystal structure of the ternary complex of eIF4E-M7GTP-4EBP2 peptide),11, 32, 34-36 we have employed extensive MD simulations to probe the stability and dynamics of eIF4E-peptide complexes. The crystal structure of the CYFIP1p-eIF4E complex is not known. To obtain a reliable model of the binding mode of CYFIP1p with eIF4E we employed advanced parallel conformational sampling algorithms,37 borrowed from methodologies developed for the estimation of protein-ligand binding free energies.38-40 Using this combined approach, we obtained what we believe is a reliable structural and energetic model of the interaction of CYFIP1p with the canonical binding site region of eIF4E. The model identifies a series of novel and unique interactions exploited by CYFIP1 to interact with eIF4E. The results of this work pave the way for alternative design strategies of peptide and peptide mimics targeting eIF4E inhibition.
Results
Cyfip1 is a noncanonical 4E binding protein
The full-length structures of eIF4E with its binding partners (4E-BPs and eIF4G) are not available. However, structures of complexes of eIF4E with 4E-BP- and eIF4G-derived peptides have been very valuable to the field.11, 34-36 In all of these examples the canonical binding sequence YXXXL
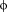 is found to be conserved among different isoforms and species [Fig. 2(A)]. Also generally conserved is the short α-helical scaffold on which the binding motif is assembled. Even though it has a very different length, structure33 and a degenerated eIF4E binding sequence, CYFIP1 has been clearly demonstrated to belong to the 4E-BP family [Fig. 2(A)].23, 26, 29 In an effort to elucidate the mode of interaction between CYFIP1p and eIF4E and compare it to those of 4E-BP- and eIF4G-derived peptides, we conducted MD simulations of these complexes.
is found to be conserved among different isoforms and species [Fig. 2(A)]. Also generally conserved is the short α-helical scaffold on which the binding motif is assembled. Even though it has a very different length, structure33 and a degenerated eIF4E binding sequence, CYFIP1 has been clearly demonstrated to belong to the 4E-BP family [Fig. 2(A)].23, 26, 29 In an effort to elucidate the mode of interaction between CYFIP1p and eIF4E and compare it to those of 4E-BP- and eIF4G-derived peptides, we conducted MD simulations of these complexes.
Based on the alignment reported in Figure 2(A) and the available set of structures of eIF4E complexes, we have selected five complexes amenable to modeling as listed in Table 1. These comprise two 4E-BP-derived peptides and three eIF4G-derived peptides.11, 34-36 Superposition of the starting conformation of the peptides shows a conserved binding mode among all isoforms, with respect to both, the positioning of the α-helix into the binding site and the orientation of the side chains of the conserved residues [Fig. 2(B)]. The complexes display substantial stability when subjected to 50 ns of MD with explicit solvation, as illustrated by the main chain RMSD trajectories, which are all within 0.3 nm of the starting structure [Fig. 2(C)]. The peptides that showed the greatest degree of conformational variation were 1RF8 and 4AZA [Fig. 2(C), yellow and violet lines]. Their distinct behavior can be rationalized by the fact that 1RF8 is an NMR derived structure34 possibly reflective of a higher degree of flexibility and 4AZA is an example of a synthetic peptide21 likely to display unique dynamics relative to the endogenous eIF4G-derived peptides. In any case, the simulation results obtained in this work for 4AZA agree with the original structural effort and modeling characterization.21 The stability of the peptides has also been analyzed in terms of persistence of secondary structure elements. In all of the cases investigated, the conformation of the helix is maintained during the MD trajectory, as indicated by the number of residues in the helix, which ranged between 4 and 8 [Fig. 2(D)].
| Parent protein | PDB ID | Organism | Peptide length | Description | Simulation length |
|---|---|---|---|---|---|
| eIF4GI | 1RF8 | S.c.a | 14 aa | MD, explicit solvent, starting from NMR model #1 | 50 ns |
| eIF4GI (D5S) | 4AZA | H.s.a | 14 aa | MD, explicit solvent, starting from crystal structure of mutated 4G-derived peptide | 50 ns |
| 4E-BP1 | 1WKW | H.s.a | 20 aa | MD, explicit solvent, starting from crystal structure | 50 ns |
| 4E-BP2 | 3AM7 | H.s.a | 19 aa | MD, explicit solvent, starting from crystal structure | 50 ns |
| eIF4GII | 1EJH | M.m.a | 16 aa | MD, explicit solvent, starting from crystal structure | 50 ns |
| CYFIP1 | 3PC8 | H.s.a | 18 aa | MD, explicit solvent, starting with CYFIP1p aligned to 4E-BP1p in 1WKW | 100 ns |
| CYFIP1 | 3PC8 | H.s.a | 18 aa | BEDAM parallel simulation, 144 replicas, starting from CYFIP1p aligned to protein-protein docking structure from Ref. 32 | 1.6 μs |
| CYFIP1 | 3PC8 | H.s.a | 18 aa | MD, explicit solvent, starting from BEDAM-predicted structure of the CYFIP1p-eIF4E complex above | 50 ns |
- a S.c., Saccharomyces cerevisiae; H.s., Homo sapiens; M.s., Mus musculus.
Based on the RMSD and helix content analysis we have selected the complex with 4E-BP1 (Table 1)35 as the most stable to employ as a starting template model for the CYFIP1p-eIF4E complex [Fig. 3(A)]. The 100 ns simulation of this complex resulted in a completely different behavior than the other peptides. This is clearly illustrated by the corresponding main chain RMSD trajectory profile [Fig. 3(B)], which is monotonically increasing without reaching a clear plateau. CYFIP1p underwent a consistent drift away from the initial conformation aligned to the 1WKW structure and, during this process, it also underwent a nearly complete loss of secondary structure, which was only partially reacquired in the last 10 ns of simulation [Fig. 3(C)].

Summary of the simulation results of the complex between CYFIP1p with eIF4E started with CYFIP1p aligned to 4E-BP in the 1WKW crystal structure. A: Superposition of the starting configuration of 1WKW and CYFIP1p. The N-terminal Leu1 and C-terminal Asn11 are indicated. B: CYFIP1 peptide all-atoms RMSD calculated as a function of time. C: Simulation time progression of CYFIP1p helix content.
The cause for both the conformational drift and unfolding are the repulsive electrostatic forces between the side chains of CYFIP1p and eIF4E residues in the initial conformation based on the structure of 4E-BP1 bound to eIF4E. Specifically, the proximity of the guanidinium group of Arg186 to the amino group of lysine at position 3 of CYFIP1p, causes an electrostatic repulsion between the two residues, as indicated by the continuous increase in the atomic distance of the centers of mass of the guanidinum and ammonium groups of the opposing residues (Supporting Information Fig. S1, gray line). At the same time a strong interaction is observed between residues Leu1 and Leu5 of CYFIP1p, establishing a hydrophobic interaction that stabilizes the new conformation of the peptide (Supporting Information Fig S1, black line). These two events together cause the unfolding of the short α-helix, the crucial secondary structure element involved in the interactions with eIF4E.
Binding of CYFIP1p to eIF4E is incompatible with the canonical binding mode of 4E-BP and eIF4G peptides
Cluster analysis of the simulation trajectories based on structural similarity was conducted to establish whether a conserved and preferred binding mode is present. The results show that for the 4E-BP and eIF4G peptides a major family of structures, containing more than 80% of total conformations sampled, can be detected (Supporting Information Fig. S2A). Furthermore, superposition of the centroids of their major clusters shows a conserved binding mode for the complexes, with the only exception of the synthetic peptide that is positioned in a slightly different conformation. Interestingly, despite undergoing unfolding, CYFIP1p is also characterized by a unique binding mode, as evidenced by the existence of a main cluster with more than 80% population (Supporting Information Fig. S2B).
In the case of 4E-BP and eIF4G peptides, the presence of the canonical sequence gives rise to a conserved profile of interaction with eIF4E (Table 2), where not only residues Y1, L6, and
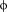 7 but also the variant residues (X2-X5, X8-X11) always interact with the same partners [Fig. 4(A) and Table 2]. Among these interactions, many are already found in the X-ray or NMR starting configurations, reported in bold in Table 2, while others appear along the simulation time due to the rearrangement of side chains (Table 2).
7 but also the variant residues (X2-X5, X8-X11) always interact with the same partners [Fig. 4(A) and Table 2]. Among these interactions, many are already found in the X-ray or NMR starting configurations, reported in bold in Table 2, while others appear along the simulation time due to the rearrangement of side chains (Table 2).

Comparison of simulated structures of the complexes of 4E-BPs and CYFIP1p with eIF4E. A: Superposition of representative snapshots showing the network of interactions established by 4E-BP and 4G peptides. B: Representative snapshot of CYFIP1p bound to eIF4E obtained by MD simulation. The side chains of eIF4E are shown in salmon, those of the peptides follow the color code defined in Figure 2.
| Peptide's residue | Position | eIF4E residue | Peptide's residue | Position | eIF4E residue | |
|---|---|---|---|---|---|---|
| 4E-BPs and eIF4G peptides | CYFIP1 peptide | |||||
| MD model | BEDAM model | |||||
| Y | Y1 | His37, Pro38, Leu39, Gly139 | L | L1 | Ile138, Gly139 | |
| D/S/G | X2 | D | X2 | |||
| R/P | X3 | Glu132, Leu135, Arg186 | K | K3 | Asp143, Asp144 | Glu132, Glu140 |
| K/E/T | X4 | R | R4 | |||
| F | X5 | His37 | L | L5 | Val69, Trp73, Ile138 | |
| L | L6 | Val69, Trp73, Leu135, Ile138, Gly139 | R | R6 | Glu140, Ser141, Asp143 | Glu132, Leu135 |
| L/M |
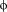 7 7 |
Trp73, Leu135 | S | S7 | ||
| D/E/G/Q | X8 | E | E8 | |||
| F/C/R | X9 | Val69, Glu70, Trp73 | C | C9 | Trp73, Leu135 | |
| K/R/Q | X10 | Trp73, Tyr76, Ile78, Glu128 | K | K10 | ||
| N/D/F | X11 | N | N11 | |||
- aInteractions conserved among MD and X-ray or NMR structures are highlighted in bold.
- Contacts occurring in more than 50% of the trajectory snapshots are listed. A contact is defined as an interatomic distance smaller than 4 Å.
The lack of conservation in the canonical sequence and the nearly complete loss of secondary structure, cause the disappearance of most of the interactions of CYFIP1p with eIF4E as observed in the simulations of the other peptides, with only 2 residues out of 11, K3, and R6, maintaining contact with eIF4E via long range electrostatic interactions [Fig. 4(B) and Table 2]. This data suggest that the starting configuration of CYFIP1p-eIF4E complex based on the 4E-BP1-eIF4E structure is not stable and it is unlikely to represent a suitable conformation for binding, making us hypothesize a different binding mode for CYFIP1p when compared with other 4E-BPs and eIF4Gs. To confirm this hypothesis we have performed binding energy distribution analysis method (BEDAM) simulations (see below and Methods) to model the formation of the CYFIP1p-eIF4E complex.
CYFIP1p interacts with eIF4E through a unique binding mode
We have employed the BEDAM to search for a likely binding mode of CYFIP1p to eIF4E. BEDAM is a well-established protocol to estimate protein-ligand binding free energies.38 One key element of the method is a hybrid potential energy function, which depends on an alchemical coupling parameter λ (see Methods). At λ = 1 the energy function represents the physical associated state of the complex whereas λ = 0 represents the uncoupled state whereby the ligand and receptor do not interact. The intermediate states at
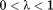 represent “alchemical” unphysical states in which receptor and ligand partially interact. This series of alchemical states composes a thermodynamic path, which connects, in a thermodynamic sense, the two end states allowing the calculation of the free energy of binding. The aim of the present calculations is to use this infrastructure, normally used to estimate binding free energies, as a conformational sampling device to locate stable bound states.39 Quantitative binding free energy estimates of eIF4E complexes, while desirable, are not yet feasible due to the long simulation times required to reach convergence.
represent “alchemical” unphysical states in which receptor and ligand partially interact. This series of alchemical states composes a thermodynamic path, which connects, in a thermodynamic sense, the two end states allowing the calculation of the free energy of binding. The aim of the present calculations is to use this infrastructure, normally used to estimate binding free energies, as a conformational sampling device to locate stable bound states.39 Quantitative binding free energy estimates of eIF4E complexes, while desirable, are not yet feasible due to the long simulation times required to reach convergence.
To maximize conformational sampling throughput, we employed not one but many MD threads in parallel41 at multiple thermodynamic states (144 threads here, see Methods). In addition to exploring conformational space, each thread is also allowed to travel in alchemical space according the so-called Hamiltonian Replica Exchange algorithm.37, 42, 43 This is an important aspect of the computational protocol and key to accelerate sampling of conformations of the complex. The transition between two stable conformations of the complex hindered by receptor-ligand contacts may be quickly achieved by a BEDAM thread that temporarily acquires a small values of λ where the ligand can move freely relative to the receptor. In this way, conformational transitions, which would normally occur on a timescale of microseconds or longer, can be made to occur on a nanosecond timescale amenable to computer simulation.44 In this application, thermodynamic states also include high temperature states.45, 46 The idea is that, while exploration in the alchemical dimension enhances sampling of intermolecular degrees of freedom (the position of the ligand relative to the receptor), access to high temperatures enhances sampling of intramolecular degrees of freedom (such as the unfolding and folding of CYFIP1p).
While the replica exchange strategy provides a level of conformational sampling unmatched by standard methods, it imposes some limitations in terms of system size.47 Accordingly, as described in detail in the Methods section, for these calculations we employ a minimal model of the eIF4E receptor (see Methods and Supporting Information Fig. S4). In addition, we imposed restraints to limit the region of conformational search. We believe that the results obtained are not significantly affected by these choices. To ensure unbiased predictions, we started the BEDAM-based conformational search by positioning the CYFIP1 peptide away from the canonical binding site of 4E-BPs. We used as a starting conformation a pose predicted by protein-protein docking experiments for full-length CYFIP132 (Supporting Information Fig. S3A) further discussed below.
This starting structure the CYFIP1 peptide, while residing on the same face of eIF4E, is significantly removed from the canonical binding site (Supporting Information Fig. S3A) and displays a set of residue-residue contact distinct from the final model discussed below. From this starting structure, the BEDAM simulations identified a unique and novel binding mode of CYFIP1p. This is within the general area of the canonical eIF4E binding site shared with 4E-BPs and eIF4Gs (Fig. 5) that caused unfolding of CYFIP1p in the brute-force MD computational experiment above. Clustering of the structures sampled by BEDAM at λ = 1 (bound state) and room temperature (T = 300 K) gave rise to 21 clusters (Supporting Information Fig. S3B). The two most populated clusters (55 and 10% of the total structures and nearly all of the structures generated in the second half of the trajectory, see Supporting Information Fig. S3B) were found to correspond to CYFIP1p bound in the canonical site of eIF4E.

Superposition of the centroids of most populated clusters found from the MD simulations of 4E-BP and 4G peptides and the BEDAM simulation of CYFIP1p complexed with eIF4E. The helix axial rotation (A) and tilt (B) of the CYFIP1 peptide (dark gray) relative to the reference 1WKW structure of 4E-BP1 are indicated. Color code as in Figure 2. C: Detail of the network of interactions established between CYFIP1p and eIF4E as predicted by the BEDAM model.
MD threads in which CYFIP1p migrated to the canonical binding site remained bound there for the remainder of the simulation where they established strong interactions with the receptor [Fig. 6(A,B)]. We think that the small fluctuations displayed in the latter part of the trajectory reflects simulation converegence and a high level of stability of this binding mode. In contrast, the BEDAM trajectories obtained using an identical procedure for three CYFIP1 mutants previously assayed23 to probe the binding hot spots of the CYFIP1-eIF4E interaction, were not observed to bind in the canonical binding site of eIF4E (results not shown). To further confirm the reliability of the structural prediction for wild-type CYFIPp, we subjected the conformation of the eIF4E-CYFIPp complex closest to the main cluster center to 50 ns to MD with explicit solvation using the same protocol used for the simulations of eIF4E-4E-BP complexes started from crystal structures above. As illustrated by the RMSD profile obtained for this system [Fig. 6(C)], the conformation of the eIF4E-CYFIPp complex remains close to the predicted binding mode for the duration of the MD run (see also Supporting Information Fig. S5). This behavior is in contrast with that observed for CYFIPp placed in the canonical binding site of eIF4E by homology with 4E-BP complexes, in which the CYFIPp peptide suffered rapid unfolding [Fig. 4(B)]. It must be therefore concluded that, according to this measure (stability within 50 ns of MD), the binding mode of the eIF4E-CYFIPp complex proposed here is as thermodynamically stable as those from the crystal structure models of 4E-BPs.

Summary of the BEDAM conformational search results for the complex between CYFIP1p and eIF4E. A: RMSD as a function of simulation time of the CYFIP1p relative to the 4E-BP peptide in the 1WKW crystal structure from the BEDAM simulation at λ = 1 and 300 K. In this simulation, CYFIP1p finds the canonical binding site after ∼4 ns of simulation and remains within it for the remainder of the simulation. B: Binding energy of the CYFIP1p-eIF4E complex as a function of simulation time for the two threads of the BEDAM simulation that contribute the most to the major population clusters at λ = 1 and 300 K temperature. In both of these threads, CYFIP1p binds eIF4E near the canonical binding site where the binding energies are most favorable. C: RMSD trajectory of the CYFIP1p peptide in the CYFIP1p-eIF4E complex during the explicit solvent simulation relative to the starting structure, which resulted from the BEDAM search (representative molecular structures are shown in Fig. S5 in Supporting Information). D: Schematic representation of the main interactions established with eIF4E by the 4E-BPs and 4Gs peptides (left panel) and CYFIP1p (right panel). The blue and red “H” stand for hydrophobic and hydrophilic residues, respectively, while “+” stand for positively charged residues.
Overall, these findings indicate that the BEDAM conformational search produced structures corresponding to the actual, stable conformational states of the complex of wild-type CYFIP1p with eIF4E. In these conformations, CYFIP1p maintains the α-helical fold conformation characteristic of the full-length protein. Furthermore, it is in a similar position and interacts with the same binding motifs of eIF4E as the 4E-BPs and eIF4G peptides. The binding mode of CYFIP1p is, never the less, distinct from those of 4E-BPs and eIF4G peptides.
Superposition of the centroid of the main cluster with those obtained for the simulations of 4E-BPs and 4Gs previously discussed, show that, relative to the consensus motif (Fig. 2), the CYFIP1 peptide is rotated on its axis counterclockwise by ∼45
 [Fig. 5(A)]. In addition it is tilted longitudinally to the protein surface by ∼20°, bringing the N-terminal end of the peptide downward along the axis of the central helix of eIF4E from the point of view of Figure 5(A,B). This novel binding mode is a direct consequence of the distinct physical characteristics of CYFIP1p, which has a degenerated canonical sequence motif. Analysis of the interactions established with eIF4E shows that CYFIP1p has a tripartite structure of interactions with the canonical binding site residues of eIF4E [Figs. 5(C) and 6(D), Table 2, and Supporting Information PDB file CYFIP1p-eIF4E.pdb]. Specifically, L5 establishes a hydrophobic cluster with Val69, Trp73, and Ile138; K3 and R6 establish an electrostatic network with Glu132 stabilized by the hydrophobic shielding of Leu135; Asn11 establishes a hydrophilic cluster with His33 and His37 [Fig. 5(C)]. These interactions are made possible thanks to the new orientation acquired by CYFIP1p and are not observed in the other peptides [Fig. 4(A) and Table 2].
[Fig. 5(A)]. In addition it is tilted longitudinally to the protein surface by ∼20°, bringing the N-terminal end of the peptide downward along the axis of the central helix of eIF4E from the point of view of Figure 5(A,B). This novel binding mode is a direct consequence of the distinct physical characteristics of CYFIP1p, which has a degenerated canonical sequence motif. Analysis of the interactions established with eIF4E shows that CYFIP1p has a tripartite structure of interactions with the canonical binding site residues of eIF4E [Figs. 5(C) and 6(D), Table 2, and Supporting Information PDB file CYFIP1p-eIF4E.pdb]. Specifically, L5 establishes a hydrophobic cluster with Val69, Trp73, and Ile138; K3 and R6 establish an electrostatic network with Glu132 stabilized by the hydrophobic shielding of Leu135; Asn11 establishes a hydrophilic cluster with His33 and His37 [Fig. 5(C)]. These interactions are made possible thanks to the new orientation acquired by CYFIP1p and are not observed in the other peptides [Fig. 4(A) and Table 2].
Discussion
The translation initiation factor eIF4E regulates protein synthesis through binding to different classes of proteins such as 4E-BP, eIF4G, and 4E-T whose binding represses or activate mRNAs translation (Fig. 1). Due to its role in controlling mRNA translation, eIF4E is involved in a wide variety of pathological processes characterized by over-expression of gene protein products, such as cancer and ASDs.1, 4-6, 8, 22 Consequently, eIF4E is a desirable target for the development of inhibitory drugs to treat these diseases. One possible strategy is to design peptides that mimic the function of 4E-BP proteins, which are the endogenous factors responsible for downregulating mRNAs expression activity.
Even though it does not share an obvious sequence and size similarity with the other members, CYFIP1 is able to repress translation of specific classes of mRNAs by binding to eIF4E, and it is therefore considered a functional member of the 4E-BPs family.23, 24, 26, 29 The mRNA inhibitory activity of CYFIP1 is strongly coupled with its concomitant regulation of actin remodeling.24, 48, 49 CYFIP1 absolves this dual role by being able, aided by a likely large conformational switch due to a particular “butterfly-like motion,” to shuttle between mRNA inhibitory complexes with eIF4E and the WAVE complex, involved in actin polymerization control.24, 32
Significantly, CYFIP1 binds eIF4E using an unique binding motif not shared with the better characterized 4E-BPs and eIF4Gs proteins. Hence, the binding mode of CYFIP1 could serve as an alternative template for the design of eIF4E inhibitor peptides. The structure of the CYFIP1-eIF4E complex is not known. Here, we have employed brute-force as well as parallel alchemical atomistic MD simulations to predict the binding mode of a CYFIP1-derived peptide (CYFIP1p) and to compare it to the known interaction profiles of 4E-BP- and eIF4G-eIF4E complexes. Collectively the results of these modeling studies show that the unusual binding sequence of CYFIP1p is not compatible with eIF4E binding in the same conformation as seen in the experimental structures of eIF4E complexed with 4E-BP and eIF4G-derived peptides. Rather, our results show that CYFIP1p adopts an unique position and orientation within the canonical binding site region of eIF4E so as to form novel specific electrostatic, hydrophobic, and hydrophilic contacts with eIF4E [Table 2 and Figs. 5(C) and 6(D)].
Previous published findings showing that mutations applied to positions 2, 3, 4, and 8 of CYFIP1p [see Fig. 2(A) for sequence numbering] result in reduced but still detectable binding23, is consistent with the present structural predictions. Of the mutations investigated, only the one at position 3 (a lysine in the wild-type sequence) is involved in direct contacts with eIF4E [Fig. 5(C)]. Hence, the binding of the mutated sequences could be accommodated in the same binding mode obtained here for wild-type CYFIP1p. The BEDAM conformational analysis for mutated CYFIP1p did not provide indications that they would bind in the canonical binding site of eIF4E like the wild-type type, so it cannot be excluded that the mutant peptides bind eIF4E in yet unidentified conformations not considered by our model.
Given the lack of crystallographic data, structural models of the interaction between the CYFIP1 full length protein and eIF4E have provided important insights on the molecular basis of FXS.23, 25 fluorescence resonance energy transfer experiments and MD simulations of full-length CYFIP1 indicate that CYFIP1 is able to switch between two different conformations based on its dual role in actin remodeling and translation control.24, 32 Protein-protein docking experiments based on this data indicate that full-length CYFIP1 is likely characterized by a binding mode similar but distinct from those experimentally observed for other 4E-BP and eIF4G peptides, and hence from the binding mode of the CYFIP1-derived peptide obtained here (Supporting Information Fig. S3A). Based on this evidence it is reasonable to hypothesize that, due to added steric hindrance and secondary contacts, the interaction pattern between full-length CYFIP1 and eIF4E could differ somewhat from that of the CYFIP1 peptide.
Overall, our findings could provide a potential alternative starting point for the design of novel and possibly more specific inhibitory peptides of eIF4E. Extensive peptide design efforts on the canonical 4E-BP and eIF4G scaffold have shown that greater inhibitory potency can be obtained by suitable set of mutations21 and by the introduction hydrocarbon staples to stabilize the α-helical motif.36 Potentially, concomitant and alternative behaviors can be accessed by taking advantage of the unique profile of interactions that characterizes CYFIP1p relative to 4E-BP and eIF4G peptides [Fig. 6(D)]. For example, CYFIP1-specific inhibitors would be less likely to produce undesirable side-effects due to unwanted activation of some mRNAs. Indeed, since 4E-BPs (eIF4E inhibitors) and eIF4Gs (eIF4E activators) share a common binding mode and establish the same network of interaction with the receptor, using their canonical sequence as a template for the design of inhibitors peptide could give rise to activation rather than inhibition of eIF4E. Accessing alternative and orthogonal modes of inhibition could serve the dual purpose of achieving additional potency as well as additional specificity.
Materials and Methods
Explicit solvent MD simulation procedure
The initial coordinates for the 4E-BPs-eIF4E and 4Gs-eIF4E complexes have been taken from X-ray or NMR derived structures (PDB ID: 1WKW, 1EJH, 3AM7, 4AZA, and 1RF8)11, 21, 34-36 (Table 1). The initial conformation of CYFIP1p has been extracted from the crystal structure of the WRC (PDB ID 3P8C),33 residues 721–734 (numbering of human protein, uniprot Q7L576) and superimposed to the structure of the Homo sapiens 4E-BP1 with code 1WKW using the Maestro program (Schrödinger, NY). Structures have been prepared using the protein preparation facility of Maestro. The complexes have been placed in a hydration box of ∼90 Å with about 20,000 TIP3P water molecules. All systems were neutralized adding the proper number of Na+ or Cl− counterions as needed. MD simulations have been conducted with the Desmond program (version 3.6, D.E. Shaw Research, NY)50 using the OPLS AA force field51 (version 2005)52 and Ewald summation for long-range electrostatic interactions on graphical processing units.
Trajectories have been collected and converted to Gromacs format to perform further analysis. Trajectory analysis has been performed with Gromacs 4.653 and in-house written code. Clustering has been performed on the main chain atoms of the peptides with the Gromos method using a cut-off of 0.17 nm and taking the structure of eIF4E as a reference.
BEDAM protocol
 (1)
(1)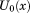 is the potential energy of the complex when receptor and ligand are uncoupled, that is as if they were separated at infinite distance from each other. The quantity b(x), the binding energy, is defined as the change in effective potential energy of the complex for bringing the receptor and ligand rigidly from infinite separation to the given conformation, x, of the complex. The system is propagated by multi-dimensional replica exchange MD37 with λ and β as exchange parameters. The purpose of the sampling along λ is to enhance mixing of conformations along the alchemical pathway while high temperatures enhance sampling of internal molecular degrees of freedom at each alchemical stage. BEDAM employs an implicit solvent representation of the system (here the OPLS/AGBNP2 model).51, 62
is the potential energy of the complex when receptor and ligand are uncoupled, that is as if they were separated at infinite distance from each other. The quantity b(x), the binding energy, is defined as the change in effective potential energy of the complex for bringing the receptor and ligand rigidly from infinite separation to the given conformation, x, of the complex. The system is propagated by multi-dimensional replica exchange MD37 with λ and β as exchange parameters. The purpose of the sampling along λ is to enhance mixing of conformations along the alchemical pathway while high temperatures enhance sampling of internal molecular degrees of freedom at each alchemical stage. BEDAM employs an implicit solvent representation of the system (here the OPLS/AGBNP2 model).51, 62In this application for each peptide we employed 144 replicas resulting from all possible combinations of 18 λ values distributed between 0 and 1 and eight temperatures distributed between 300 and 417 K. Replica exchange was conducted using the Asynchronous Replica Exchange software module (https://github.com/ComputationalBiophysicsCollaborative/AsyncRE) running on the Brooklyn College WEB computational grid. Periodically replicas were allowed to exchange λ and β parameters, allowing each replica to move in conformational space as well as alchemical and temperature space. The average simulation period between parameter exchanges was set to 25 ps. Conformations were saved on disk according to the same period. On average, each replica underwent ∼450 exchange periods resulting in 10 ns of simulation time per replica on average or ∼1.5 μs of overall simulation time per peptide. The conformations and binding energies of the replicas at λ = 1 and T = 300 K at each time point were extracted and analyzed.
To enhance computational throughput the BEDAM parallel conformational search employed a reduced model of the eIF4E receptor (Supporting Information Fig. S4). Specifically, we modeled only the canonical binding site region and regions around it composed of residues 68 − 77, 122 − 142, and 185 − 189 of eIF4E. The C-α atoms of this construct were restrained by a spherically symmetric harmonic potential with force constant 0.3 kcal/mol
 Å2. The dangling ends of simulated chains were capped with standard N-methylacetamide and acetyl capping groups. The CYFIP1-derived peptides were not restrained with the exception of a flat-bottom harmonic potential tether centered at 8 Å distance with 8 Å tolerance between the C-α atom of Glu132 of eIF4E and the C-α atom of Lys725 of CYFIP1 [position 3 in Fig. 2(A)]. The imposition of a restraining tether between the ligand and receptor is required by the alchemical statistical mechanics theory on which BEDAM is based.54, 55, 63 The size of the tolerance parameter has been adjusted to minimize its effect.
Å2. The dangling ends of simulated chains were capped with standard N-methylacetamide and acetyl capping groups. The CYFIP1-derived peptides were not restrained with the exception of a flat-bottom harmonic potential tether centered at 8 Å distance with 8 Å tolerance between the C-α atom of Glu132 of eIF4E and the C-α atom of Lys725 of CYFIP1 [position 3 in Fig. 2(A)]. The imposition of a restraining tether between the ligand and receptor is required by the alchemical statistical mechanics theory on which BEDAM is based.54, 55, 63 The size of the tolerance parameter has been adjusted to minimize its effect.
Conclusions
CYFIP1 is an eIF4E binding protein (4E-BP) and, similarly to other known 4E-BPs, acts as repressor of mRNA translation by binding to eIF4E and preventing the formation of actively translating polysomes. Unwanted activation of eIF4E leads to unchecked cellular proliferation and cancer. Dysregulation of eIF4E has also been linked to the insurgence of ASDs and FXS. The study of the mechanism of inhibition of eIF4E by 4E-BPs is key to the development of drug therapies targeting this pathway. The parallel MD results obtained in this work indicate that CYFIP1p interacts with eIF4E by means of an unique binding mode not shared by other known 4E-BP proteins. These findings elucidate the mechanism by which CYFIP1p interacts with eIF4E despite having a sequence binding motif significantly different from the established canonical binding motif of most 4E-BP and can serve as a starting point for alternative eIF4E inhibitor peptides design strategies, possibly leading to constructs with superior potency and specificity than currently available. Further studies should be directed towards the biochemical validation of the structural predictions here described and to address in vivo the relevance of the recently discovered secondary lateral binding site of eIF4E in the context of the CYFIP1-eIF4E interaction.
Acknowledgments
Computations were conducted at the Computational Molecular Biophysics Laboratory at the Department of Chemistry at the Brooklyn College of the City University of New York. The BEDAM parallel simulations were conducted on the WEB computational grid of Brooklyn College; the authors thank Sade Samlalsingh, James Roman and John Stephen, William Flynn and Junchao Xiao, for assistance with the grid software and infrastructure and VP Mark Gold for making the student PC laboratory computational facilities of Brooklyn College available for research. The authors dedicate this work to Ronald M. Levy, a pioneer of biomolecular modeling and simulation.
References
Abbreviations
-
- 4E-BP
-
- eIF4E-binding protein
-
- BEDAM
-
- binding energy distribution analysis method
-
- CYFIP1
-
- cytoplasmic Fmrp interacting protein 1
-
- CYFIP1p
-
- CYFIP1 peptide
-
- eIF4E
-
- eukaryotic translation initiation factor 4E
-
- eIF4G
-
- eukaryotic translation initiation factor 4 gamma
-
- FMRP
-
- fragile X mental retardation protein
-
- MD
-
- molecular dynamics
-
- RMSD
-
- root mean square deviation