Changes in Protein Glycosylation as a Result of Aptamer Interactions with Cancer Cells
Abstract
Purpose
Based on the recent aptamer-related breast cancer studies, which indicate the therapeutic role of specific oligonucleotide sequences, experiments have been designed in an attempt to unravel the molecular targets of this mechanism. This article describes the study on glycoproteome changes in breast cancer cells as a result of their interactions with aptamers.
Experimental design
Aberrations in protein glycosylation play an important role in tumorigenesis and influence cancer progression, metastasis, immunoresponse, and chemoresistance, therefore this study is focused on the identification of the alterations in glycan expression on the surface of proteins as a potential and innovative tool for biomedical applications of aptamers in cancer treatment.
Results
Two proteins, kinesin-like protein (KI13B) and proliferating cell nuclear antigen (PCNA), have been identified that carry N-glycan epitopes after conjugation with aptamer sequences.
Conclusions and clinical relevance
Multiple features of aptamers as an alternative to protein antibodies are utilized for various biomedical applications ranging from biomarker discovery, bioimaging, targeted therapy, drug delivery, and drug pharmacokinetics and biodistribution. Frequently, aptamers bind to their target molecules and modulate their function. Such therapeutic aptamers can modify the biological pathways for treatment of many types of diseases, such as cancer.
1 Introduction
Aptamers are short oligonucleotides with unique 3D structures selected to bind to a target of interest with high affinity and specificity. This feature is used by the Nature for regulation of RNA functions (riboswitches), where aptamers interact with a variety of molecules. Aptamers are produced during chemical synthesis, therefore they can be modified in many ways, including addition of linkers, immobilization on the surface of nanomaterials, as well as attaching fluorophores and dyes. In comparison to antibodies, they are effortless to obtain, and consequently are characterized by a low cost of production with no batch to batch variations. Because of their properties, aptamers have been increasingly exploited for diverse applications, including biomarker discovery, regulation of gene expression, specific target isolation, biosensors, imaging, and diagnostics.1 Moreover, aptamers often represent the ability to change the expression level of their target proteins, thus they can act as therapeutics.2 Several aptamers have been engaged in clinical trials, and couple of them is already FDA approved and utilized as drugs.3 Their application in theranostics, which combines diagnosis and therapy, might give a possibility to provide more specific, individualized treatment and reduction of side effects for patients.
Synthetic aptamers are selected among the vast set of oligonucleotide sequences (libraries) during the process called SELEX (Systematic Evolution of Ligands by Exponential Enrichment).4 During cell-SELEX procedure, a library of sequences, usually 1015, is incubated with target cells. Subsequently, oligonucleotides bound to the target are recovered and amplified by PCR or RT-PCR (Figure 1). Oligonucleotides that were not attached to the target are removed. Successively, the reduced pool of sequences is used for further rounds of cell-SELEX. Moreover, the pool is verified during incubation with control cells and, opposite to the target cells, bound fraction is rejected, as only sequences with interactions specific to the target cells are preferred. The cell-SELEX process usually takes 10–15 cycles to obtain specific aptamer.

Therapeutic function of aptamers is of a great interest, as they can change the expression level of their target proteins.2 Aptamers interacting directly with cancer cells for cancer therapy can modulate the immune system and indirectly inhibit cancer cell growth.3 However, the mechanisms underlying the aptamer-mediated anti-proliferation effect have not been fully understood.1
In this study, we have identified the glycoproteomic changes in breast cancer cells as a result of their interactions with therapeutic aptamers. We have also validated protein N-glycosylation pattern in MCF7 cells after incubation with aptamers, revealing their potential importance as a tool for biomedical applications in cancer treatment.
Clinical Relevance
Although aptamers represent a promising class of drugs and drug-delivery systems, their use in therapeutics and biomolecular medicine is relatively new. Only one sequence is approved for the therapeutic purpose (Macugen) and few are in clinical trials. Nonetheless, the number of preclinical studies using aptamer has increased significantly in recent years giving hope for their application as a new treatment method. With this study, we present two aptamers that might be potential therapeutics and theranostics in breast cancer. KI13B and PCNA proteins were identified as aptamer protein targets that were characterized by altered glycosylation profile. Subsequently, lack of glycan epitopes was observed in control and scrambled sequence samples. As a result, it might be speculated that the protein glycosylation occurs as a result of aptamer sequence binding followed by protein conformation changes.
In conclusion, the use of aptamers to develop therapeutic agents and to understand the possible mechanism of its function is still in its infancy, and more advanced toxicity study should be carried, but the already achieved successful preclinical results are very promising.
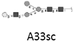
MCF7 cells were selected as a most popular breast cancer model of human mammary gland adenocarcinoma. The procedure of aptamer selection specific for MCF7 cells was described elsewhere.5 A series of experiments were performed, where target cells were incubated with two aptamers, A26 and A33, and a scrambled sequence, A33sc, described as inert to MCF7 cells.5 The capillary LC combined with MS/MS approach was designed for proper protein recognition.6 Prior to MS-based analysis, proteins were enriched in N-glycosylated fraction using PHA-L (Phaseolus vulgaris Leucoagglutinin Lectin) lectin affinity chromatography, which is widely described as characteristic for the highly branched N-glycans that are present in many different cancerous cells, and subsequently separated using gel electrophoresis.7
2 Experimental Section
2.1 Cell Culture
Breast cancer cells MCF7, purchased form Sigma–Aldrich ECACC (The European Collection of Authenticated Cell Cultures), were cultured in RPMI 1640 (Sigma–Aldrich) supplemented with 10% Fetal Bovine Serum FBS (Life) and 1% antibiotic/antifungal solution (Sigma–Aldrich) at 37 °C in 5% CO2 humidified atmosphere.
2.2 Aptamers Binding Assay
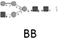
Aptamers (Microsynth AG) stored at –20 °C were thawed to the room temperature. Stock solutions (25 mm in DPBS buffer) of the oligonucleotide sequences were incubated at 90 °C for 5 min, and then cooled down to room temperature by the natural cooling process to the 37 °C facilitating the proper 3D structures of aptamers during renaturation. Oligonucleotides were diluted in binding buffer (BB; 5 mm MgCl2, 1 mm CaCl2, 0.1% ssDNA in DPBS [Sigma–Aldrich]) to a final concentration of 25 nm. Cells were incubated at 37 °C for 45 min with the aptamer sequences A26 and A33 that were found to be highly specific toward MCF7 cells,8 and with A33sc (scrambled sequence) characterized by the lack of specificity to the studied cells (Table 1). Additional control sample was prepared after incubation with BB only. After binding, the excess of aptamers was removed for 2 min with 5 mL of washing buffer (5 mm MgCl2, 1 mm CaCl2, 1 mg mL–1 BSA in DPBS) on rotator at 37 °C and the process was repeated three times. Washing step did not affect viability of the cells, nor binding kinetics between cells and aptamers. The cells were carefully scraped off the flask surface, transferred into a 15 mL Falcon tube, and centrifuged at 200 × g for 3 min. All experiments were repeated three times as three independent biological replicates. After the cells have formed a monolayer on the surface of the T75 dish (Eppendorf), the passage was performed in proportion 1:3. Cells were grown until the proper amount of biological material necessary to complete the experiments was obtained.
| Aptamer | Sequence |
|---|---|
| A26 | 5′biotin-GCTGTGTGACTCCTGCAACCGGACTGCAGAGACCGTCTGTCGGTGAACACTATTAGACGCGGCAGCTGTATCTTGTCTCC-3′ |
| A33 | 5′biotin-GCTGTGTGACTCCTGCAAGGCGGTACGCGTGTGGACAGAAGTGACCGCCAATAGCGCCTGGCAGCTGTATCTTGTCTCC-3′ |
| A33sc | 5′biotin-GCTGTGTGACTCCTGCAAGACGGACCAGAGGGCGGAGAGCTTTGGCAGCTCTCGGCATCAAGCAGCTGTATCTTGTCTCC-3′ |
2.3 Homogenization
MCF7 cells were homogenized three times by sonication on ice (5 s each) in DPBS buffer with the proteinase inhibitors cocktail (Roche). Homogenates were centrifuged for 45 min at 25 000 × g at 4 °C. Protein concentration was measured using Bradford method (Sigma–Aldrich), according to the producer's protocol.
2.4 Glycoprotein Enrichment
For the identification of N-glycosylation profiles, PHA-L (Vector Laboratories) was selected. PHA-L specifically binds to the variety of highly branched-type N-glycans that are characteristic for many tumor cells.7 Therefore, MCF7 homogenates were incubated with 50 µL of PHA-L lectin linked to agarose for 1 h at room temperature in the presence of 300 µL of 10 mm HEPES buffer, pH 7.5, with 0.15 m NaCl, 0.1 mm CaCl2, and 0.01 mm MnCl2 (all from Sigma–Aldrich), followed by 16 h incubation at 4 °C. Successively, suspensions were centrifuged at 25 000 × g for 5 min at 4 °C. Precipitates were washed three times with 1 mL of PBS (Sigma–Aldrich) followed by 5 min incubation at 95 °C with 40 µL of the Laemmli sample buffer (Bio-Rad; protein concentration 250 µg).
2.5 Electrophoretic Separation
1D electrophoresis was performed as follows. Proteins were separated with the aid of the 10% SDS-PAGE in a Mini Protean system (Bio-Rad). The gels were stained with the Sypro Ruby (Thermo Fisher Scientific) for total protein visualization, and the ProQ Diamond (Thermo Fisher Scientific) prior to glycoprotein identification, followed by the nanoLC–MS/MS analysis. Electrophoretic separations were run individually for each biological replicate. Gel scans were performed using the XR+ GelDoc (Bio-Rad).
2.6 Isolation of Peptides
The spots corresponding to glycoproteins visualized by the ProQ Diamond were excised from the gel with a scalpel, chopped into cubes, rinsed with water, and transferred into the LoBind tubes (Eppendorf). Dye was removed with 100 mm NH4HCO3 (Sigma–Aldrich), and an equal volume of 100% acetonitrile (ACN; JT Baker) was added after 10 min. Then, the gel pieces were treated with 100% ACN and re-swollen in 5 ng mL–1 trypsin (Promega) in 50 mm NH4HCO3 for 45 min on ice. The supernatants, which were not absorbed by gel particles, were removed and gel pieces were immersed in 50 mm NH4HCO3 and incubated overnight at 37 °C. After completion of digestion, the supernatants were transferred into fresh tubes, followed by the addition 100 µL of 50 mm NH4HCO3, and after 10 min, an equal volume of 100% ACN was added. The samples were incubated under shaking for 15 min at 37 °C. Extraction of peptides was repeated twice with 5% formic acid (FA; Sigma–Aldrich) (v/v) in 100% ACN, and the combined extracts were evaporated to dryness in a vacuum centrifuge. The resulting peptides were dissolved in 20 µL of 0.1% FA.9
2.7 NanoLC–MS/MS Analysis
The nano liquid chromatography – tandem mass spectrometry (nanoLC–MS/MS) analysis was performed with the aid of the Proxeon capillary chromatography system combined with the AmaZon ETD mass spectrometer (Bruker Daltonics).10 The separation was performed in a capillary column filled with the PepMap reversed-phase material (C18, 15 cm long, 75 µC;m ID, 3 µm particle size, 100 Å pore size [Thermo Fisher Scientific]). The gradient was formed using 0.1% FA in water (solvent A) and 0.1% FA in ACN (solvent B), and it was delivered at a flow rate of 300 nL min–1. The system was controlled by the Hystar software (Bruker Daltonics). A gradient was produced from 2% to 50% B in 30 min and up to 90% B at 35 min, then kept until 45 min, and again reduced to 2% until 55 min. The AmaZon ETD instrument operated in a positive-ion mode. During analysis, two most intense peaks (threshold above 100 000) in the range 450–1800 m/z were automatically fragmented using CID in the data-dependent acquisition mode. The acquired spectra were analyzed using the Bruker Data Analysis 4.0 software and were identified using the Mascot 2.4.1 algorithm against the Swiss-Prot sequence database 2018_03 (557012 sequences; 199714119 residues). Search parameters were set as follows: taxonomy, human; modification, carbamidomethyl (fixed); methionine dioxidation (variable), up to 1 missed cleavage; peptide charges, +1, +2, and +3; mass tolerance, 0.8 Da for precursor mass and 0.6 Da for fragment mass. Proteins with more than two fragmented peptides detected were considered, and an additional criterion was an ion score above 40. Accumulation time to achieve the number of ions entering the trap was less than 1 ms. Analysis of glycopeptides was performed using the GlycoQuest software (Bruker Daltonics). Search parameters were set as follows: minimal precursor m/z, 600 Da; occurrence of oxonium ions (IonTrapOxoniumIons List) with absolute mass tolerance, 0.3 Da; minimum intensity coverage, 50%; distance-based glycopeptides pattern established with the minimal number of consecutive m/z distances required, 2; MS tolerance, 0.3 Da; MS/MS tolerance, 0.35 Da; peptide mass offset, 204.09 Da.
2.8 Western Blotting
Verification of the MS-identified protein sequences was performed with standard anti-KI13B and anti-PCNA protein antibodies. Moreover, affirmation of the N-glycan attachment to studied proteins was achieved by using the biotinylated PHA-L form, followed by detection with alkaline phosphatase-conjugated streptavidin.
To confirm aptamers binding to their protein targets, we have also used alkaline phosphatase-conjugated streptavidin to detect biotinylated forms of aptamers on the surface of identified proteins. Standard proteins could not be used, because of the lack of information on their glycosylation state, therefore MCF7 cell homogenates after biotin-conjugated aptamers incubation were applied, followed by lectin affinity enrichment.
Blotted membranes, along with the Bio-Rad standard protocol, were treated with casein solution (Vector Laboratories) for 1 h at room temperature on rotator, washed three times with Tris buffered saline TBS (Sigma–Aldrich), and subsequently incubated with anti-KI13B (Thermo Fisher Scientific), anti-PCNA (Thermo Fisher Scientific), and biotinylated PHA-L lectin for 2 h, according to the manufacturer's recommendations. After washing with TBS buffer, the blots were incubated with (Vector Laboratories) for 2 h with TBS supplemented with 0.1% Tween 20. Finally, the blots were visualized by alkaline phosphatase substrate kit II (Vector Laboratories). Blots were documented with the XR+ GelDoc (Bio-Rad).
3 Results and Discussion
MCF7 cells after incubation with aptamer sequences were homogenized, and protein extracts were enriched in N-glycan fraction with PHA-L chromatography, followed by electrophoretic separation.
3.1 Protein Profile Changes
As a result of SDS-PAGE separation, three gels were obtained that were first stained with a ProQ Diamond dye characteristic for glycoproteins, followed by staining with SyproRuby for total protein visualization (Figure 2).

Confirmation of the aptamer–protein target complexes formation was achieved by the Western blotting method, using biotinylated forms of aptamers, followed by the detection with alkaline phosphatase-conjugated streptavidin (Figure 3).

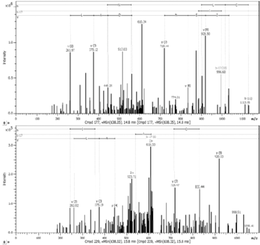
Seven fractions of N-glycoproteins in PHA-L enriched pull-outs were excised and analyzed using capillary LC–MS/MS. It was possible to identify 522 proteins in all types of samples A26, A33, A33sc, and BB (Table S1, Supporting Information).
Fourteen proteins were prevalent in the samples incubated with both aptamers A26 and A33 that were not present in control samples A33sc and BB (Figure 4). Functional characteristics of these proteins were presented in Figure 5.


In the group of identified proteins, there are molecules of TRAP1 Heat shock protein 75 kDa responsible for nucleotide binding, protein folding, response to stress, and apoptosis signaling pathway,11 likewise a membrane component responsible for protein localization and trafficking VP13A (Vacuolar protein sorting-associated protein 13A).12 Also, a co-chaperone, which acts as a regulator of the Hsp70 chaperone machinery that may be involved in the processing of ataxia-linked proteins SACS (Sacsin), has been identified in samples interacting with aptamers A26 and A33.13 Furthermore, a protein that plays an important role in the regulation of stress-induced apoptosis UACA (Uveal autoantigen with coiled-coil domains and ankyrin repeats) has been found in the samples incubated with A26 and A33 aptamers.14 Essentially, all proteins present in the MCF7 protein extracts after incubation with potential therapeutic aptamers sequences and not identified in control samples were described in the literature as cancer-related agents.15, 16
3.2 Glycoproteome Changes
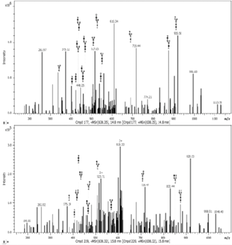
Finally, using MS approach, it has been recognized, for the first time, the presence and localization of the specific N-glycan in the amino acid sequences among identified proteins (Table 2). Four proteins with altered N-oligosaccharides profiles were identified; two of them characteristic for the A26 sequence KI13B and PCNA, and one for the A33 oligonucleotide ANXA2. Also, one protein HS90B was found to be deglycosylated as a result of A26 and A33 interactions.
| Protein name | Peptide sequence | Peptide spectrum | Glycan spectrum | Glycan structure/sample |
|---|---|---|---|---|
| Kinesin-like protein KI13B |
R.TFVNGSSVSSPIQLHHGDR.I 566 AA |
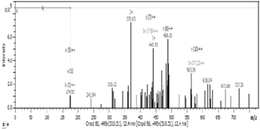 |
 |
 |
| Proliferating cell nuclear antigen PCNA |
R.NLAMGVNLTSMSK.I 121 AA |
 |
 |
 |
| Annexin A2 ANXA2 |
K.GVDEVTIVNILTNR.S 130 AA |
 |
 |
 |
| Heat shock protein HSP 90-beta HS90B |
R.ELISNASDALDK.I 96 AA |
 |
 |
  |
The protein identified in our study as uniquely glycosylated is KI13B (Kinesin-like protein KIF13B), which is a phosphoprotein responsible for the reorganization of cytoskeleton, intracellular trafficking, microtubule-based movement, protein targeting, and nucleotide binding.17 Mass of this molecule is more than 200 kDa, and so far, it was localized in the cell interior; however, the presence of the N-glycan on the 566 amino acid residue might bring a light on the new locus of KI13B on the membrane surface.
The presence of N-glycosylated KI13B in A26 sample was confirmed using Western blot analysis in two-step immunodetection; first using antibodies characteristic for their polypeptide sequences, and second occurrence of highly complex antennary N-oligosaccharides by the biotinylated anti-PHAL antibody (Vector Laboratories; Figure 6).

Another protein found to be N-glycosylated is PCNA (Proliferating cell nuclear antigen) that plays a key role in DNA damage response, controls eukaryotic replication, cell proliferation, and epithelial cell differentiation, and was identified in A26 samples. The presence of PCNA proteins with attached N-glycan was confirmed using Western blot approach (Figure 7). Glycosylation of this protein at the 121 AA position may influence its localization and cause cancerous transformation by changing cell differentiation and proliferation mechanisms.18

Annexin 2 ANXA2 was described as an aptamer-binding target in MCF7 cells.8 This membrane-bound protein that is involved in heat stress response, angiogenesis, cell proliferation, and adhesion may cross the membrane as a constituent of the lipid raft assembly; however, it has not been described to occur in the glycosylated form. Using MS-based approach, we have identified, for the first time, the structure of N-glycan at the 130 asparagine residue of Annexin 2 in A33 samples.
HS90B (Heat shock protein HSP 90-beta) is a molecular chaperone that promotes, along with its co-chaperones, the maturation, folding, and proper regulation of specific target proteins involved in cell cycle control, differentiation, gene expression, transcription factors, and signal transduction.19 It was found to be N-glycosylated at the 96 arginine residue in control samples, while the lack of glycan was observed in MCF7 samples incubated with aptamers A26 and A33.
In summary, the strategy described in this work enabled efficient discovery of N-glycosylation patterns and their precise localization in cancerous cells as a result of aptamers interactions.
The presented approach represents an effective identification method to reveal aptamer–protein target interactions that may influence their glycosylation profile. We believe that after formation of aptamer–protein conjugates, the 3D structure is changed, and steric effect might therefore alter the action of glycotransferases, which in turn have an impact on the protein glycosylation state. Changes of the glycosylation profile occur at different stages in diseased cells. The detection of these changes might be critical for understanding the molecular mechanisms underlying pathogenesis, as well as for diagnosing disease states and monitoring therapeutic effects. Aptamers have been shown as candidates for molecular theranostics agents simultaneously serving as therapeutic and diagnostic tools.20
Acknowledgements
The manuscript was written through contributions of all authors. All authors were given approval to the final version of the manuscript. This study is supported by Grant “META” No. 5/EuroNanoMed/2012, in addition to the Polish Ministry of Science and Higher Education 2013/09/B/NZ4/02531, and 2016/21/B/NZ6/01307.
Conflict of Interest
The authors declare no conflict of interest.