

Structure-based design of diarylpyrimidines and triarylpyrimidines as potent HIV-1 NNRTIs with improved metabolic stability and drug resistance profiles
Abstract
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are an important component of anti-acquired immunodeficiency syndrome treatment regimen. In the present work, with the previously reported compound K-16c as lead, a series of novel 2,4,5-trisubstituted pyrimidine derivatives were designed based on the cocrystal structure of K-16c/RT, with the aim to improve the anti-human immunodeficiency virus type-1 (HIV-1) activities and metabolic stability properties. Compound 11b1 exhibited the most potent antiviral activity against wild-type (WT) and a panel of single mutant HIV-1 strains (EC50 = 2.4−12.4 nM), being superior to or comparable to those of the approved drug etravirine. Meanwhile, 11b1 exhibited moderate cytotoxicity (CC50 = 4.96 μM) and high selectivity index (SI = 1189) toward HIV-1 WT strain. As for HIV-1 RT inhibition test, 11b1 possessed excellent inhibitory potency (IC50 = 0.04 μM) and confirmed its target was RT. Moreover, the molecular dynamics simulation was performed to elucidate the improved drug resistance profiles. Moreover, 11b1 was demonstrated with favorable safety profiles and pharmacokinetic properties in vivo, indicating that 11b1 is a potential anti-HIV-1 drug candidate worthy of further development.
1 INTRODUCTION
Acquired immunodeficiency syndrome (AIDS), mainly caused by the human immunodeficiency virus type-1 (HIV-1),1 is a serious epidemic disease posing a significant threat to public health worldwide.2 According to the data from the World Health Organization, approximately 39.0 million individuals are currently living with HIV, including 1.3 million new infections reported in 2022.3 In the viral replication cycle of HIV-1, reverse transcriptase (RT) has become the preferred target for the development of anti-HIV-1 agents due to its well-solved structure and definite mechanism of action.4, 5 RT inhibitors include nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs). NNRTIs bind to an allosteric site known as the NNRTI binding pocket (NNIBP), located approximately 10 Å away from the polymerase active site, which could induce conformational changes in the catalytic domain of RT, thereby disrupting the viral reverse transcription process.6, 7 Therefore, NNRTIs have gained an important position in the development of anti-HIV-1 drugs due to their prominent antiviral efficacy and high specificity.
To date, six NNRTIs have been approved by the US Food and Drug Administration, including first-generation drugs nevirapine (NVP), delavirdine (DLV), and efavirenz (EFV), as well as second-generation drugs etravirine (ETR), rilpivirine (RPV), and doravirine (DOR).8, 9 However, the lack of intrinsic exonucleolytic proofreading during RT reverse transcription of HIV-1 lead to its low genetic barrier, resulting in the rapid development of drug resistance.10 For example, the single-mutant strains K103N and Y181C frequently occurred in first-generation NNRTIs therapy,11 whereas single-mutant strain E138K and double-mutant strains F227L + V106A and K103N + Y181C (RES056) were observed with second-generation NNRTIs.12, 13 Moreover, serious adverse reactions including hepatotoxicity, severe rash, and hypersensitivity reactions also limited their clinical application.14, 15 Therefore, there is an urgent need to develop novel NNRTIs with higher efficiency, improved drug resistance profiles, and lower toxicity.
Previous efforts in our group have prompted the identification of 2,4,5-trisubstituted pyrimidine compound K-16c (Figure 1), which exhibited enhanced anti-HIV-1 potency and drug resistance profiles compared to that of ETR.16 The cocrystal structure revealed that extensive hydrophobic interactions and a network of backbone hydrogen-bonding interactions formed between K-16c and NNIBP contribute to its improved drug resistance profiles (Figure 2).17 However, K-16c suffered from a metabolic stability challenge related to a short half-life (T1/2 = 1.74 h). Metabolite analysis of previously discovered effective compounds indicated that the piperidine-linked methylene motif proved to be the readily metabolizable moiety, resulting in the relatively short half-life of K-16c (T1/2 = 1.74 h).18 To further improve anti-resistance profiles and drug-like properties, we first retained the privileged central scaffold of the lead K-16c, while removing the metabolic-perturbing methylene group on the right-wing. Meanwhile, various substituted phenyl groups were introduced on the piperidine ring to further explore the chemical space of NNIBP and enrich the structure–activity relationships (SAR) in the solvent-exposed region (Figure 2).


2 RESULTS AND DISCUSSION
2.1 Synthesis of the target compounds
The synthetic protocols for the newly designed derivatives are outlined in Scheme 1-4. As shown in Scheme 1, the starting material 4-(N-Boc-amino)piperidine (1) was treated with 4-fluoronitrobenzene and 3-fluoronitrobenzene to obtain 2a and 2b, which were removed the tert-butoxycarbonyl protecting group to yield the key intermediates 3a and 3b, respectively. As depicted in Schemes 2, 2,4-dichloropyrimidine (4) was reacted with 4-hydroxy-3,5-dimethylbenzonitrile and (E)-3-(4-hydroxy-3,5-dimethylphenyl) acrylonitrile to give intermediates 5a and 5b, which were undergone nucleophilic substitutions with 3a and 3b to obtain intermediates 6a1−2 and 6b1−2, respectively.19 Treatment of 6a1−2 and 6b1−2 with NIS to prepare 7a1−2 and 7b1−2. Finally, the corresponding target compounds 8a1−8 and 8b1−8 were obtained through the Suzuki reaction, reduction reaction, and acylation reaction.20 The reaction conditions for intermediates 10a−c are similar to that of 3a−b, with the exception that the substituents on the benzene ring. Then, intermediates 5a−b underwent nucleophilic substitution reaction with 10a−c to obtain target compounds 11a1−3 and 11b1−3.16




2.2 Anti-HIV activity evaluation of target compounds 8a1−8 and 8b1−8
The antiviral activity of all target compounds were evaluated in MT-4 cell lines infected with HIV-1 wild-type (WT) strain (IIIB) and NNRTIs-resistant strain RES056 by the MTT method.21 The FDA approved NNRTIs drugs NVP, EFV, and ETR were used as references. The cellular biological evaluation results, expressed as EC50 value (antiviral activity), CC50 value (cytotoxicity), and SI value (selectivity index, CC50/EC50 ratio).
As shown in Table 1, compounds 8a1−8 and 8b1−8 of Series I exhibited effective potency against HIV-1 IIIB strain, with EC50 values ranging from 4.0 to 48.6 nM, which were superior to that of NVP (EC50 = 180 nM). Among them, the introduction of the NHSO2CH3 substituent on the right benzene ring proved to be optimal for antiviral activity, as exemplified by compounds 8a3 (EC50 = 5.5 nM), 8a7 (EC50 = 5.5 nM), 8b3 (EC50 = 6.4 nM), and 8b7 (EC50 = 4.7 nM). Replacing the substituents position from para- to meta- (8a3 vs. 8a7 and 8b3 vs. 8b7) further enhanced the antiviral activity. Notably, compounds 8a6 (EC50 = 4.0 nM) exhibit the most effective HIV-1 IIIB inhibitory activity, being more potent than that of DOR (EC50 = 8.5 nM), but weak to that of RPV (EC50 = 1.0 nM) and ETR (EC50 = 2.9 nM). When the cyano (CN) group in the left wing was replaced with a cyanovinyl (CV) group, the yielded compounds showed significantly increased activity against the HIV-1 IIIB and RES056, indicating that the cyanovinyl group was more beneficial to improve the activity than the cyano group. Except for the compound 8b1 (EC50 = 2895 nM), all other CV-substituted compounds exhibited nanomolar level activity against RES056 (EC50 = 33.5−830 nM). Especially, 8b6 (EC50 = 33.5 nM) showed the most active potency, being superior to that of DOR (EC50 = 128 nM) and comparable to that of ETR (EC50 = 35.4 nM). Moreover, 8b6 exhibited decreased cytotoxicity (CC50 = 15.55 μM) and higher selectivity (SI = 464 and 1969) compared with EFV and ETR.
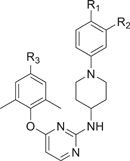 |
||||||||
| Compds | R1 | R2 | R3 | EC50 (nM)a | CC50 (μM)b | SIc | ||
|---|---|---|---|---|---|---|---|---|
| IIIB | RES056 | IIIB | RES056 | |||||
| 8a1 | NO2 | H | CN | 21.8 ± 4.5 | ≤913 | 9.13 ± 1.70 | 419 | >418.7 |
| 8a2 | NH2 | H | CN | 48.6 ± 10.2 | 1821 ± 621 | 21.68 ± 3.12 | 446 | 12 |
| 8a3 | NHSO2CH3 | H | CN | 5.5 ± 1.7 | 516 ± 160 | 29.27 ± 4.14 | 5302 | 57 |
| 8a4 | NHSO2NH2 | H | CN | 26.8 ± 13.3 | 1990 ± 954 | 23.84 ± 1.05 | 892 | 12 |
| 8a5 | H | NO2 | CN | 9.5 ± 4.9 | 3044 ± 1321 | 70.57 ± 8.44 | 7463 | 23 |
| 8a6 | H | NH2 | CN | 4.0 ± 0.75 | 221 ± 93.7 | 26.72 ± 4.86 | 6629 | 121 |
| 8a7 | H | NHSO2CH3 | CN | 5.5 ± 2.8 | 373 ± 53.1 | 12.55 ± 1.30 | 2268 | 34 |
| 8a8 | H | NHSO2NH2 | CN | 36.5 ± 27.7 | 1017 ± 126 | 27.45 ± 1.50 | 753 | 27 |
| 8b1 | NO2 | H | CV | 10.6 ± 7.1 | 2895 ± 3253 | 36.89 ± 4.34 | 3472 | 13 |
| 8b2 | NH2 | H | CV | 30.9 ± 9.8 | 532 ± 218 | 9.97 ± 3.20 | 322 | 19 |
| 8b3 | NHSO2CH3 | H | CV | 6.4 ± 1.9 | 97.0 ± 45.7 | 37.64 ± 11.89 | 5845 | 388 |
| 8b4 | NHSO2NH2 | H | CV | 16.5 ± 8.1 | 247 ± 31.8 | 15.19 ± 2.17 | 917 | 61 |
| 8b5 | H | NO2 | CV | 13.9 ± 9.1 | 830 ± 546 | 15.56 ± 1.86 | 1116 | 19 |
| 8b6 | H | NH2 | CV | 7.9 ± 2.4 | 33.5 ± 7.8 | 15.55 ± 5.93 | 1969 | 464 |
| 8b7 | H | NHSO2CH3 | CV | 4.7 ± 1.1 | 62.5 ± 27.6 | 7.15 ± 0.65 | 1505 | 114 |
| 8b8 | H | NHSO2NH2 | CV | 7.4 ± 1.6 | 178 ± 54.5 | 7.90 ± 1.24 | 1063 | 44 |
| NVP | 180 ± 63 | >1502 | >15.02 | >86 | NA | |||
| ETR | 2.9 ± 0.48 | 35.4 ± 14.2 | >4.61 | >1616 | >130 | |||
| EFV | 3.3 ± 0.83 | 415 ± 127 | >6.35 | >1928 | >15 | |||
| RPV | 1.0 ± 0.27 | 10.7 ± 7.96 | 3.98 | 3989 | 371 | |||
| DOR | 8.5 ± 3.6 | 128 ± 60.6 | >294 | >34 588 | >2297 | |||
- Abbreviations: DOR, doravirine; EFV, efavirenz; ETR, etravirine; HIV-1, human immunodeficiency virus type-1; NVP, nevirapine; RPV, rilpivirine.
- a EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytopathicity, as determined by the MTT method.
- b CC50: concentration required to reduce the viability of mock-infected cell cultures (cytotoxicity, CC) by 50%, as determined using the MTT method.
- c SI: selectivity index, the ratio of CC50/EC50.
Then, these potent compounds (8a6−7, 8b3, and 8b6−8) against HIV-1 IIIB and RES056 were selected to evaluate their activity against other common single-mutations (L100I, K103N, Y181C, Y188L, and E138K) and double-mutation (F227L + V106A). As shown in Table 2, all the selected compounds showed low EC50 values, ranging from 6.0 to 331 nM, which were superior to NVP and EFV, but inferior to ETR and RPV. Notably, the most promising compound 8b6 showed a 1.6- to 12.1-fold decrease in activity against all mutant strains compared to that of ETR. We speculate that the decreased activities may be due to the combined effect of introducing a pyridine ring in regulatory region II and the right wing directly connected benzene ring, resulting in conformational change of the compounds and decreased affinity with mutant RT.
| Compds | EC50 (nM)a | |||||
|---|---|---|---|---|---|---|
| L100I | K103N | Y181C | Y188L | E138K | F227L + V106A | |
| 8a6 | 28.9 ± 16.2 | 6.0 ± 2.6 | 29.1 ± 9.4 | 48.6 ± 3.6 | 12.4 ± 1.6 | 89.1 ± 9.8 |
| 8a7 | 71.0 ± 6.5 | 6.6 ± 2.0 | 15.2 ± 2.9 | 253 ± 87.2 | 39.2 ± 3.7 | 331 ± 90.2 |
| 8b3 | 20.6 ± 10.1 | 7.5 ± 2.7 | 43.6 ± 7.5 | 52.4 ± 5.6 | 40.0 ± 31.1 | 25.0 ± 22.8 |
| 8b6 | 7.4 ± 3.5 | 6.0 ± 2.4 | 31.0 ± 24.5 | 47.4 ± 26.3 | 37.6 ± 31.3 | 107 ± 84.0 |
| 8b7 | 9.1 ± 3.4 | 14.4 ± 15.9 | 35.6 ± 19.1 | 54.0 ± 22.9 | 35.1 ± 26.7 | 69.1 ± 19.3 |
| 8b8 | 23.3 ± 16.2 | 10.9 ± 4.0 | 51.5 ± 11.0 | 62.7 ± 17.4 | 57.2 ± 11.2 | 166 ± 67.2 |
| NVP | 931 ± 627 | 6248 ± 3284 | 9025 ± 2621 | >748 | 129 ± 52.7 | >748 |
| ETR | 4.5 ± 2.5 | 2.9 ± 0.55 | 12.8 ± 4.7 | 21.4 ± 14.0 | 7.1 ± 2.1 | 8.8 ± 5.1 |
| EFV | 23.7 ± 9.1 | 51.6 ± 16.7 | 8.7 ± 3.9 | 244 ± 88.3 | 5.9 ± 1.7 | 139 ± 87.3 |
| RPV | 1.54 ± 0.00 | 1.31 ± 0.36 | 4.73 ± 0.48 | 79.4 ± 0.77 | 5.75 ± 0.11 | 81.6 ± 21.2 |
| DOR | 13.3 ± 9.8 | 13.9 ± 4.8 | 26.8 ± 7.1 | 2084 ± 1697 | 37.4 ± 15.1 | 67 849 ± 25 160 |
- Abbreviations: DOR, doravirine; EFV, efavirenz; ETR, etravirine; HIV-1, human immunodeficiency virus type-1; NVP, nevirapine; RPV, rilpivirine.
- a EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytopathicity, as determined by the MTT method.
2.3 Molecular modeling studies of 8b6
To gain a more specific explanation of the reduced potency against resistant strains, 8b6 was further conducted molecular modeling analysis, to further explore its binding modes in the RT allosteric pocket. The molecular dynamics simulations were performed with the docked 8b6-RT complexes during 200 ns.22-24 The RMSD (root mean square deviation) plot (Supporting Information S1: Figure 1) illustrated that the binding of ligand-RT conformations were steady throughout 200 ns simulation, which proved that compound 8b6 was stabilized in the allosteric site of HIV-1 RT. Then, the conformations of 8b6 in HIV-1 WT and mutant RTs were investigated. As shown in Figure 3, the left-wing of 8b6 maintained hydrophobic interactions with surrounding residues Y181, Y188, and W229. Besides, the pyrimidine ring well accommodated the entrance channel consisting of residues E138, V179, I180, and Y181, which could form water-bridges with the backbone carbonyl of E138 and I180. In WT, L100I, and K103N RTs, 8b6 adapted the NNIBP with the classical “U”-shaped conformation, and maintained hydrogen bonds of NH linker and the backbone of K101. However, In Y181C, F227L + V106A, and RES056 RTs, the conformations of the 8b6 were deviated, and lost the key hydrogen bonding between NH and K101, which rationalize the decreased activity of 8b6 against these mutant strains.25

In addition, MMGBSA calculations were used to determine the relative binding strengths of 8b6 to WT RT and different mutant RTs. As shown in Table 3, the binding affinity of 8b6 with WT, L100I, and K103N HIV-1 RTs was higher compared to those of Y181C, Y188L, E138K, and F227L + V106A HIV-1 RTs. All the binding energy components altogether culminate into the overall MMGBSA, which provided a reasonable explanation for the sharply decreased potency against the Y181C, Y188L, E138K, and F227L + V106A mutant strains and in support of its biological evaluation results (EC50 = 31.0−107 nM).
| MMGBSA (kcal/mol) | WT | L100I | K103N | Y181C | Y188L | E138K | F227L + V106A | K103N + Y181C |
| −110.21 | −112.73 | −112.55 | −100.68 | −105.40 | −104.26 | −92.20 | −99.99 |
- Abbreviation: WT, wild-type.
2.4 Anti-HIV activity evaluation of target compounds 6a1−2, 6b1−2, 11a1−3, and 11b1−3
According to the molecular docking results in Figure 3, due to steric hindrance caused by the bulky molecular volume, the conformations of 8b6 were deviated and failed to maintain the “U”-shaped conformation in the complexes of Y181C, F227L + V106A, and RES056 RT with 8b6, thus losing the critical hydrogen bonds between NH linker and K101 backbone. Structural simplification is defined as the reduction of molecular complexity and molecular size. To reduce molecular obesity of the newly designed 2,4,5-trisubstituted pyrimidine derivatives in Series I, structural simplification by removing non-essential groups represents an effective strategy during the lead optimization. Therefore, in continuation of our exploration, the replacement of the bulky pyridine ring with a hydrogen atom was conducted via the structural simplification strategy, with the expectation of maintaining the classical “U”-shaped conformation within NNIBP and improving the drug resistance profiles against mutant strains. In addition, diverse substituents harboring hydrogen bond donors/acceptors were further introduced on the right-wing phenyl moiety, yielding diarylpyrimidine compounds 6a1−2, 6b1−2, 11a1−3, and 11b1−3 (Series II). As shown in Table 4, all compounds exhibited nanomolar activity against HIV-1 IIIB with EC50 values of 2.3−28.1 nM, which were increased by twofold compared to that of the triarylpyrimidines. More encouragingly, 11b1 (EC50 = 78.9 nM) and 11b3 (EC50 = 68.0 nM) also exhibited effective antiviral activity against RES056, being much better than that of EFV (EC50 = 244 nM) and DOR (EC50 = 128 nM). Meanwhile, 11b1 and 11b3 also endowed with moderate cytotoxicity (CC50 = 4.96 and 6.59 μM), and sufficient SI values (SI = 1189 and 2024) toward HIV-1 WT strain. The preliminary SAR of substituents could be summarized as below: R3 substituent is CV, the activity was significantly increased compared to CN; the activity order of R1 and R2 substituents was SO2CH3≥CONH2>SO2NH2>m-NO2>p-NO2.
| Compounds | R1 | R2 | R3 | EC50 (nM)a | CC50 (μM)b | SIc | ||
|---|---|---|---|---|---|---|---|---|
| IIIB | RES056 | IIIB | RES056 | |||||
| 6a1 | NO2 | H | CN | 28.1 ± 14.5 | >7970 | 7.97 ± 1.49 | 284 | ≤1 |
| 6a2 | H | NO2 | CN | 4.7 ± 1.1 | >7740 | 10.94 ± 2.95 | 2314 | ≤1 |
| 6b1 | NO2 | H | CV | 4.2 ± 0.36 | 26 755 ± 14 660 | >265.83 | >62 500 | >10 |
| 6b2 | H | NO2 | CV | 8.7 ± 2.6 | 449 ± 291 | 2.59 ± 0.51 | 296 | 6 |
| 11a1 | SO2CH3 | H | CN | 6.6 ± 3.0 | 903 ± 165 | 6.27 ± 0.51 | 949 | 7 |
| 11a2 | SO2NH2 | H | CN | 8.2 ± 0.89 | 3249 ± 608 | 22.16 ± 2.43 | 2717 | 7 |
| 11a3 | CONH2 | H | CN | 18.4 ± 20.8 | 936 ± 126 | 7.83 ± 0.81 | 426 | 8 |
| 11b1 | SO2CH3 | H | CV | 4.2 ± 1.1 | 78.9 ± 19.5 | 4.96 ± 0.82 | 1189 | 63 |
| 11b2 | SO2NH2 | H | CV | 2.3 ± 0.75 | 115 ± 47.7 | 10.42 ± 3.78 | 4471 | 91 |
| 11b3 | CONH2 | H | CV | 3.2 ± 2.1 | 68.0 ± 37.3 | 6.59 ± 0.57 | 2024 | 97 |
| NVP | 211 ± 98.0 | >1502 | >15.02 | >71 | NA | |||
| ETR | 3.5 ± 0.98 | 36.6 ± 15.5 | >4.61 | >1299 | >126 | |||
| EFV | 5.3 ± 3.1 | 244 ± 122 | >6.35 | >1190 | >26 | |||
| RPV | 1.00 ± 0.27 | 10.7 ± 7.96 | 3.98 | 3989 | 371 | |||
| DOR | 8.5 ± 3.6 | 128 ± 60.6 | >294 | >34 588 | >2297 | |||
- Abbreviations: DOR, doravirine; EFV, efavirenz; ETR, etravirine; HIV-1, human immunodeficiency virus type-1; NVP, nevirapine; RPV, rilpivirine.
- a EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytopathicity, as determined by the MTT method.
- b CC50: concentration required to reduce the viability of mock-infected cell cultures by 50%, as determined by the MTT method.
- c SI: selectivity index, the ratio of CC50/EC50.
Furthermore, these compounds were evaluated for their activity against mutant strains. As displayed in Table 5, the most promising compound 11b1 showed effective efficacy against all tested mutant strains, with EC50 values of 5.8 nM (L100I), 2.4 nM (K103N), 10.7 nM (Y181C), and 12.4 nM (E138K), being superior to that of ETR in the same cellular assay, with the exception of Y188L (EC50 = 286 nM). Especially, 11b1 (EC50 = 10.1 nM) showed effective efficacy against the highly resistant double-mutant strain F227L + V106A, exhibiting about 2-, 8-, and up to 6718-fold activity enhancement over that of ETR (EC50 = 22.4 nM), RPV (EC50 = 81.6 nM), and DOR (EC50 = 67849 nM), respectively, and was proved to be the most effective inhibitor among all the newly synthesized compounds.
| Compds | EC50 (nM)a | |||||
|---|---|---|---|---|---|---|
| L100I | K103N | Y181C | Y188L | E138K | F227L + V106A | |
| 6a1 | 1533 ± 136 | 58.1 ± 11.7 | 852 ± 168 | >7970 | 678 ± 238 | 483 ± 83.3 |
| 6a2 | 385 ± 77.6 | 29.6 ± 24.6 | 91.1 ± 39.9 | 6561 ± 1443 | 132 ± 24.5 | 809 ± 329 |
| 6b1 | 20.3 ± 13.0 | 8.6 ± 0.15 | 20.3 ± 6.6 | 106 793 ± 10 145 | 41.7 ± 21.0 | 565 ± 777 |
| 6b2 | 61.6 ± 42.4 | 26.5 ± 14.9 | 60.4 ± 32.5 | 1130 ± 371 | 70.0 ± 39.5 | 414 ± 49.6 |
| 11a1 | 306 ± 73.8 | 9.7 ± 1.5 | 84.3 ± 22.8 | 1465 ± 60.7 | 113 ± 74.3 | 32.8 ± 21.6 |
| 11a2 | 536 ± 227 | 12.7 ± 2.5 | 160 ± 50.9 | 3012 ± 344 | 143 ± 48.6 | 175 ± 128 |
| 11a3 | 127 ± 24.8 | 5.8 ± 2.1 | 63.9 ± 7.1 | 989 ± 340 | 51.2 ± 22.9 | 65.6 ± 8.1 |
| 11b1 | 5.8 ± 2.1 | 2.4 ± 0.0 | 10.7 ± 1.9 | 286 ± 68.7 | 12.4 ± 3.1 | 10.1 ± 3.6 |
| 11b2 | 20.2 ± 16.0 | 2.4 ± 0.96 | 14.9 ± 3.5 | 701 ± 348 | 11.6 ± 1.0 | 55.9 ± 7.9 |
| 11b3 | 41.8 ± 30.1 | 4.8 ± 3.5 | 47.1 ± 38.7 | 471 ± 112 | 14.8 ± 4.7 | 72.9 ± 13.6 |
| NVP | 1310 ± 1378 | 4864 ± 3703 | 2196 ± 820 | >748 | 157.8 ± 54.7 | >7.48 |
| ETR | 7.2 ± 7.2 | 2.9 ± 1.6 | 16.1 ± 5.7 | 21.9 ± 9.2 | 8.1 ± 1.3 | 22.4 ± 17.2 |
| EFV | 23.2 ± 13.9 | 77.5 ± 35.8 | 7.2 ± 2.8 | 260 ± 80.5 | 8.2 ± 4.2 | 193 ± 77.8 |
| RPV | 1.54 ± 0.00 | 1.31 ± 0.36 | 4.73 ± 0.48 | 79.4 ± 0.77 | 5.75 ± 0.11 | 81.6 ± 21.2 |
| DOR | 13.3 ± 9.8 | 13.9 ± 4.8 | 26.8 ± 7.1 | 2084 ± 1697 | 37.4 ± 15.1 | 67 849 ± 25 160 |
- Abbreviations: DOR, doravirine; EFV, efavirenz; ETR, etravirine; HIV-1, human immunodeficiency virus type-1; NVP, nevirapine; RPV, rilpivirine.
- a EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytopathicity, as determined by the MTT method.
2.5 Molecular modeling studies of 11b1
To investigate the binding stability and dynamic behavior of 11b1 with HIV-1 RT, the 200 ns MD simulation of 11b1-RT complexes was further conducted. The RMSD plot (Suppoting Informaton S1: Figure 2) showed the binding conformations were steady, indicating the strong binding between 11b1 and RT. The binding modes of 11b1 in WT and mutant RTs indicated that compound 11b1 could accommodated well into the NNIBP in a typical “U”-shaped conformation (Figure 4), exhibiting several key pharmacophore features and protein−ligand interactions: (i) the NH linker of 11b1 developed the key hydrogen bond with the K101 main-chain that is essential for maintaining binding affinity; (ii) the cyanovinyl motif on the left-wing stretched into the hydrophobic tunnel formed by residues Y181, Y188, F227, and W229, forming π−π stacking interactions with these aromatic residues; (iii) the pyrimidine-N atom could form hydrogen-bonding interactions with the E138 side-chain through a water bridge molecule; (iv) the right-wing of the piperidine-linked phenyl motif occupied the solvent-exposed region consisting of residues V106, Y318, and P236, and was involved in the extensive hydrogen bond network. However, the substitution of Y188 to L188 leads to a significant reduction in the π−π stacking interactions between the left-wing benzene ring and Tyr188, which are necessary to improve binding affinity. Besides, the Y188L mutation resulted in a change in the binding pocket, where spatial conflict between L188 and Y181 shifted the original “face-to-face” angle between Y181 and the benzene ring, which may further explain the significantly reduced activity of 11b1 against the Y188L strain compared to other resistant strains. As shown in Table 6, except for the 11b1-Y188L RT complex, 11b1 has a high affinity for binding to WT, L100I, K103N, Y181C, E138K, F227L + V106A, and RES056 RTs. The MD simulation results demonstrated that 11b1 can maintain a good conformation in various RTs while retaining key protein−ligand interactions, which reasonably explained the excellent antiviral activity of 11b1.

| MMGBSA (kcal/mol) | WT | L100I | K103N | Y181C | Y188L | E138K | F227L + V106A | K103N + Y181C |
| −103.94 | −98.94 | −104.35 | −96.34 | −93.81 | −96.30 | −97.72 | −97.89 |
- Abbreviations: HIV-1, human immunodeficiency virus type-1; WT, wild-type.
2.6 HIV-1 RT inhibition assay
To further confirm the binding target of these novel derivatives, 8b3, 8b6−7, and 11b1−3 were selected to evaluate their ability to inhibit the WT HIV-1 RT enzyme,26 and the results were summarized in Table 7. Compounds 11b1−3 (IC50 = 0.024−0.040 μM) showed a 1.8- to 6.1-fold increase in activity compared to those of 8b3 and 8b6−7 (IC50 = 0.11−0.17 μM), and exhibited comparable inhibitory activity to that of ETR (IC50 = 0.013 μM). Overall, the RT inhibition results validated that these novel derivatives have high binding affinity for RT and belong to the typical HIV-1 NNRTIs.
| Compounds | IC50 (RT) (μM)a | Compounds | IC50 (RT) (μM)a |
|---|---|---|---|
| 8b3 | 0.15 ± 0.01 | 11b1 | 0.040 ± 0.002 |
| 8b6 | 0.17 ± 0.03 | 11b2 | 0.033 ± 0.003 |
| 8b7 | 0.11 ± 0.01 | 11b3 | 0.024 ± 0.00 |
| NVP | 0.40 ± 0.10 | ETR | 0.013 ± 0.002 |
| RPV | 0.015 ± 0.001 | DOR | 0.019 ± 0.002 |
- Abbreviations: DOR, doravirine; ETR, etravirine; HIV-1, human immunodeficiency virus type-1; NVP, nevirapine; RPV, rilpivirine; WT, wild-type.
- a IC50: inhibitory concentration required to inhibit biotin deoxyuridine triphosphate incorporation into WT HIV-1 RT by 50%.
2.7 Physicochemical properties assessment
To further investigate the preliminary drug-likeness of novel diarylpyrimidine derivatives, the physicochemical properties of representative compounds 11b1−3 were characterized by using the software ADMETlab 2.0. As shown in Table 8, the hydrogen bond acceptor (nHBAcc), hydrogen bond donor (nHBDon), rotatable bond (nROTB), and octanol-water partition coefficients (Clog P) of these three compounds are consistent with the “Lipinski's rule of five”, while ETR and RPV (Clog P = 5.22 and 5.75) deviates slightly from the acceptable range. In addition, we calculated the topological polar surface area (TPSA) of the compounds to estimate their absorption and membrane permeability. The TPSA values of compounds 11b1 and 11b3 are 116.59 and 93.37 Å2, indicating that they have the advantage of oral absorption (<140 Å2) and are unlikely to cross the blood-brain barrier (>60 Å2). Meanwhile, compounds 11b1−3 have significantly increased Fsp3 values of 0.26−0.30 compared to that of ETR and RPV.
| Parameter items | Acceptable range | 11b1 | 11b2 | 11b3 | ETR | RPV |
|---|---|---|---|---|---|---|
| MW | <500 Da | 503.62 | 504.6 | 468.55 | 435.29 | 366.43 |
| nHBAcc | <10 | 6 | 7 | 5 | 7 | 6 |
| nHBDon | <5 | 1 | 2 | 2 | 3 | 2 |
| nROTB | <10 | 7 | 7 | 7 | 4 | 5 |
| Clog P | <5 | 4.10 | 3.33 | 3.56 | 5.22 | 5.75 |
| TPSA | ≤140 Å2 | 116.59 | 142.61 | 93.37 | 120.65 | 97.42 |
| Fsp3 | 0.30 | 0.27 | 0.26 | 0.10 | 0.09 |
- Abbreviations: Clog P, octanol-water partition coefficient; ETR, etravirine; Fsp3, fraction of sp3 carbon atoms; Fsp3, sp3-hybridized carbon atoms/total carbon atoms; MW, molecular weight; nHBAcc, number of hydrogen bond acceptors; nHBDon, number of hydrogen bond donors; nROTB, number of rotatable bonds; RPV, rilpivirine; TPSA, topological polar surface area.
2.8 In vivo acute toxicity assessment
Acute toxicity experiments were conducted on 11b1 using Kunming healthy mice. Twenty Kunming mice were divided into two groups: one group were received 2000 mg/kg of 11b1 orally, and the other group were served as the control. In the next 7 days, there were no abnormal phenomena or deaths in the mice in the experimental group. As shown in Figure 5, the drug administration group showed no significant difference in body weight gain compared to the blank control group.

2.9 In vivo subacute toxicity assessment
Next, the subacute toxicity of 11b1 was further investigated in healthy Kunming mice to determine its in vivo safety evaluation. After 2 weeks of intragastric administration of 11b1 at a dose of 50 mg/kg every other day, no mortality and no signs of toxicity (lethargy, clonic convulsion, anorexia, and ruffled fur) were observed in the mice of the 11b1-treated group. Furthermore, the organ toxicity experiment was examined by hematoxylin−eosin (HE) staining. As depicted in Figure 6, no apparent physiological abnormalities or lesions were observed in the heart, liver, spleen, lungs, and kidneys, suggesting the higher safety profiles of 11b1.

2.10 In vivo pharmacokinetic profiles of 11b1
The results of in vivo pharmacokinetic evaluation of 11b1 was shown in Table 9 and Figure 7. After intravenous injection of 2 mg/kg in Sprague−Dawley rats, 11b1 showed a modest clearance rate (CL = 52.6 mL/min/kg), the maximum concentration (Cmax) and AUC0-t of 11b1 were 1897 ng/mL and 653 h/ng/mL. After an oral dose of 10 mg/kg, the Cmax of 11b1 was reached at 0.5 h with a value of 36.6 ng/mL. Although the half-life of 11b1 (T1/2 = 2.23 h) was significantly improved over that of K-16c (T1/2 = 1.74 h), its bioavailability (F) was only 3.88%, which needs further improvement for an ideal drug candidate.
| Subject | T1/2 (h) | Tmax (h) | Cmax (ng/mL) | AUC0-t (h/ng/mL) | AUC0-∞ (h/ng/mL) | CL (mL/min/kg) | F (%) |
|---|---|---|---|---|---|---|---|
| 11b1 (iv)b | 0.698 ± 0.038 | 0.0333 ± 0.00 | 1897 ± 416 | 653 ± 148 | 659 ± 149 | 52.6 ± 13.0 | |
| 11b1 (po)c | 2.23 ± 0.424 | 0.500 ± 0.00 | 36.6 ± 14.5 | 116.4 ± 40.38 | 127.7 ± 40.02 | 3.88 |
- a PK parameter (mean ± standard deviation, n = 3).
- b Dosed intravenously at 2 mg/kg.
- c Dosed orally at 10 mg/kg.

3 CONCLUSION
Herein, guided by the available structural biology information, a series of novel diarylpyrimidines and triarylpyrimidines derivatives were rationally designed and synthesized, to enhance the metabolic stability properties and improve the drug resistance profiles. The biological evaluation results demonstrated that most of the compounds yielded excellent inhibitory activity against HIV-1 IIIB and mutant strains. Among them, 11b1 turned out to be the most promising inhibitor, with EC50 values of 4.2 nM (IIIB), 5.8 nM (L100I), 2.4 nM (K103N), 10.7 nM (Y181C), 12.4 nM (E138K), 10.1 nM (F227L + V106A), and 78.9 nM (RES056), being superior to or comparable to those of ETR (EC50 = 2.9−36.6 nM) and EFV (EC50 = 5.3−244 nM) in the parallel cellular assay, with the exception of Y188L (EC50 = 286 nM). More importantly, 11b1 exhibited improved in vivo metabolic stability (T1/2 = 2.23 h) and safety properties (LD50 > 2000 mg/kg). Taken together, this work contribute to the discovery of 11b1 as a potent HIV-1 NNRTIs with promising antiviral activities and druggability profiles for further development.
AUTHOR CONTRIBUTIONS
Erik De Clercq and Christophe Pannecouque performed anti-HIV potency in vitro. Fabao Zhao performed molecular dynamic simulation analysis. Zhao Wang, Peng Zhan, Xinyong Liu, and Dongwei Kang are the administrators of the study, they reviewed and edited the manuscript. Minghui Xie, Ye Li, Zongji Zhuo, and Xin Li performed the design, synthesis, and structure confirmation of these compounds. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from the National Natural Science Foundation of China (82273773, 81973181), National Key Research and Development Program (2023YFE0206500), Major Basic Project of Natural Science in Shandong Province (ZR2021ZD17), the Foreign Cultural and Educational Experts Project GXL20200015001), the Qilu Young Scholars Program of Shandong University, and the Taishan Scholar Program of Shandong Province.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
Open Research
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.

